→ Definition: Beim Morbus Wilson handelt es sich um eine autosomal-rezessiv, vererbte Störung des Kupferstoffwechsels; Ausgangpunkt hierbei ist ein Defektes der Transport-ATPase für Kupfer in den Leberzellen. In der Folge kommt es zu einer verminderten biliären Kupferausscheidung und reduzierten Synthese von Coeruloplasmin, wodurch sich wiederum eine Überladung von Cu2+ in den verschiedenen Geweben wie Leber, Gehirn, Augen, Nieren und weiteren parenchymatösen Organen entwickelt. Der Morbus Wilson manifestiert sich vor allem in der Leber (mit z.B. chronischer Hepatitis, Leberzirrhose, akutem Leberversagen), Gehirn (z.B. Stammgangliensymptomatik), am Augen (Kornea als Kayser-Fleischer-Kornealring) und den Erythrozyten (Hämolyse).
→ Epidemiologie: Insgesamt handelt es sich bei der Wilson-Krankeit weltweit gesehen um eine seltene Erkrankung:
→ I: Die homozygote Form findet man in 1/200000 Fällen, die heterozygote Form 1/200.
→ II: Das Erkrankungsalter liegt > 6. Lebensjahr mit initialer Lebermanifestation; in Ausnahmefällen entwickelt sich die Wilson-Krankheit auch jenseits des 40. Lebensjahres.
→ III: Ab dem 10. Lebensjahr treten häufig zusätzliche Symptome z.B. neurologische Störungen (Beteiligung der Basalganglien) auf.
→ Ätiologie: Es zeigt sich eine Mutation des Wilson-Gens (= ATP-7B-Gen) auf Chromosom 13q14,3, welches das Wilsonprotein, eine P-Typ-ATPase (= Kupfertransportprotein) kodiert. Das Protein ist für die Translokation von Kupfer im Golgi-Apparat und endoplasmatischen Reticulum verantwortlich.
→ Pathophysiologie: Das über die Nahrung aufgenommene Kupfer (2-5mg/d) wird zur Leber transportiert und dort gespeichert, biliär ausgeschieden oder an Coeruloplasmin (> 95%) gebunden. Beim Morbus Wilson findet man eine verminderte biliäre Ausscheidung. Normalerweise wird über die Galle 2mg Kuper pro Tag ausgeschieden, beim Wilson liegt die Ausscheidungsrate nur bei 0,2-0,4mg/d. Zudem zeigt sich eine verminderte Konzentration an Coeruloplasmin, sodass die Gesamtkonzentration des Kupfers im Serum, sowie die gebundene Kupferkonzentration vermindert ist. Charakteristischerweise ist hierbei das freie zytotoxisch-wirkende Kupfer im Serum erhöht und lagert sich in der Leber und den anderen Geweben vermehrt an.
→ Klinik: Die Krankheit manifetiert sich nicht vor dem 6. Lebensjahr, sondern tritt zumeist in der 2. Lebensdekade selten nach der 4 Lebensdekade auf;
→ I: Frühsymptome: Hepatomegalie mit flüchtigem Ikterus, Anämie sowie evtl. Leuko- und Thrombozytopnie.
→ II: Hepatische Manifestation: Asymptomatisch nur mit Transaminansenerhöhung, über die Fettleber bis hin zur fulminant verlaufenden Hepatitis mit akutem Leberversagen. Das Endstadium stellt die Leberzirrhose mit ihren Komplikationen z.B. portale Hypertension, hepatische Enzephalopathie, Varizenblutungen etc. dar.
→ IÍI: Augensymptome: Charakteristisch sind insbesondere:
→ 1) Der Kayser-Fleischer-Kornealring, eine goldbraune Verfärbung des Kornealrandes, durch Ablagerung von Kupfer in der Descemet-Membran der Kornea.
→ 2) Sowie der Sonnenblumenkatarrakt.
→ IV: Blut: Eine Hämolyse tritt beim Morbus Wilson zumeist in Verbindung mit einem akuten Leberversagen auf. Ursache ist die Hemmung von Enzymen der Glykolyse als Folge einer Kupferüberladung der Erythrozyten. Häufig besteht zugleich auch eine Koagulopathie. Thrombozytopenie und Leukozytopenie werden aufgrund einer Knochenmarkschädigung durch Kupfer hervorgerufen.
→ V: Neurologische Symptome: Die klinische Symptomatik ist bei ZNS-Manifestation sehr variabel. Verhaltensauffälligkeiten, Störungen beim Schlucken und Sprechen sind oftmals die ersten Symptome. Da die Kupferablagerung zumeist in den Stammganglien erfolgt, betreffen die neurologischen Störungen insbesondere die Motorik (extrapyramidal-motorisch) mit Koordinationsstörungen, Tremor, Rigor, Dyskinesien, vermehrtem Speichelfluss, bei Kleinhirnbeteiligung Ataxie, Dysarthrie und Nystagmus. In 20% der Fälle ist die neurologische Symptomatik mit
→ VI: Psychiatrischen Auffälligkeiten: Wie Wesensveränderung, Konzentrationsschwäche, kognitive Störungen, depressive Verstimmung, Stimmungslabilität (insbesondere läppischer Euphorie), Angst- und Panikattacken, aber auch katatone Psychosen bis hin zur Demenz vergesellschaftet.
→ VII: Weitere Organsymptome:
→ 1) Tubuläre Nierenschädigung: Mit Proteinurie, Glukosurie, Nierensteinen und Nephrosklerose (sekundäres Fanconi-Syndrom)
→ 2) Herzbeteiligung: Kardiomyopathie und Rhythmusstörungen.
→ 3) Gelenkbeteiligung durch Einlagerung von Kalziumpyrophosphatdihydrat im Rahmen einer Chondrokalzinose.

→ Klassifikation: Der Morbus Wilson lässt sich anhand des Krankheitsverlaufes klassifizieren in:
→ I: Frühsymptome: Hepatomegalie, intermittierende ikterische Schübe, Hämolyse, Anämie, Leuko- und Thrombozytopenie.
→ II: Hepatische Form: Ausbildung von Hepatitiden mit Progredienz einer postnekrotischen Zirrhose und evtl. Leberversagen.
→ III: Hepatozerebrale Verlaufsform: Man findet Symptome wie Tremor, Rigor, Dysarthrie (Störung des Sprechens), Hypomimie, Hypersalivation, sowie läppische Euphorie, psychischer Abbau sowie Leberbeteiligung.
→ IV: Neurologische Verlaufsform: Hierbei kommt es zu progredienten neurologischen Ausfällen mit Intensitätstremor und unkoordinierten Bewegungsabläufen, skandierender Sprache (verlangsamte, abgehackte, verwaschene Sprache) und zunehmender Hilflosigkeit.
→ Diagnose: Patienten mit unklarer Leberenzymerhöhung und unklaren neurologischen Symptomen sollte immer an einen Morbus Wilson gedacht werden.
→ I: Augen: Charakteristischerweise Nachweis des Kayser-Fleischer-Kornealrings in der Spaltlampenuntersuchung.
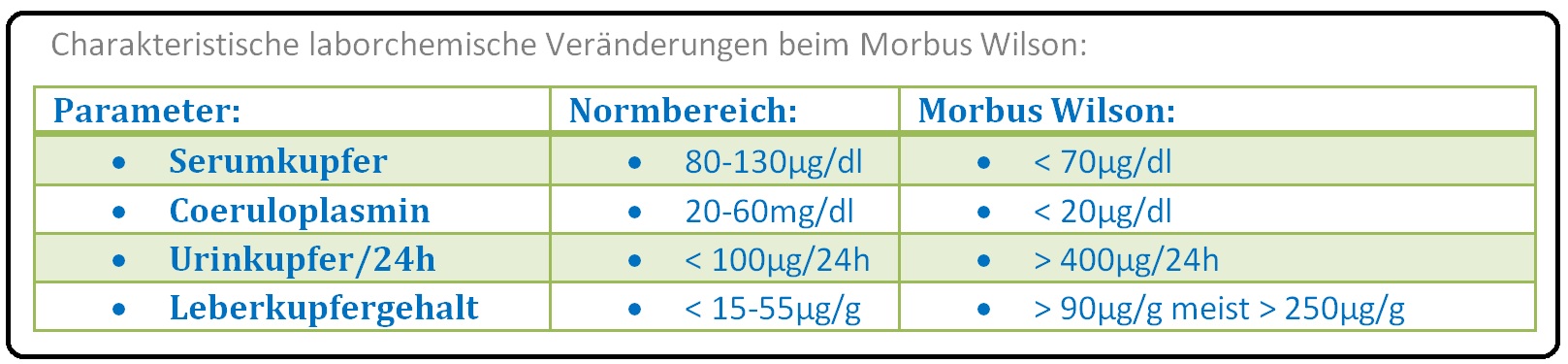
→ II: Labor:
→ 1) Gesamt-Kupfer im Serum (80-130µg/dl): < 70µg/dl.;
→ 2) Freies Kupfer im Serum: > 10µg/dl,
→ 3) Kupfer im 24h-Sammelurin (< 100µg/24h): Meist > 400µg/24h;
→ 4) Coeruloplasminbestimmung (geringe Sensitivität, hohe Spezifität; Coeruloplasmin ist ein Syntheseparameter der Leber sowie ein Akut-Phaseprotein; 20-60mg/dl): < 20 mg/dl, zumeist < 15mg/dl.
→ 5) Kupfergehalt der Leber (< 15-55µg/g Trocken-Leber): > 90 µg/g, meist sogar 250µg/g im Biopsat.
→ III: Leberhistologie:
→ 1) Frühstadium: Mit mitochondrialen Veränderungen, Verfettung und Mallory Bodies.
→ 2) Spätstadium: Fibrotische und zirrhotische Leberveränderungen.
→ IV: Klinische Testverfahren:
→ 1) D-Penicillamin-Belastungstest: Nach Gabe von D-Penicillamin kommt es zur deutlichen Steigerung der Kupferausscheidung im 24h-Urin.
→ 2) Radioaktiv-Kupfertest: Nach Gabe eines radioaktiven 64-Cu2+ kommt es im Serum normalerweise zu einem 2-gipfeligen Anstieg der Radioaktivität. Charakteristischerweise fehlt der 2. Gipfel beim Morbus Wilson, da er den Einbau des markierten Cu2+ in das Coeruloplasmin darstellt.
→ 3) Genetischer Nachweis der Mutation des Wilson Gens.
→ Differenzialdiagnose: Hiervon abzugrenzen sind u.a.:
→ I: Weitere hepatische Erkrankungen wie nicht-alkoholbedingte Steatohepatitis (= NASH), Hepatitis anderer Genese, pimär sklerosierende Cholangitis und die primär biliäre Zirrhose.
→ II: Erniedrigte Coeruloplasmin-Werte bestehen auch beim nephrotischen Syndrom, bei der exudativer Enteropathie, Malabsoprtionssyndrom, Malnutrition (Mangelernährung).
→ Klinisch-relevant: Bei unklaren Lebererkrankungen vor dem 35. Lebensjahr stets an den Morbus Wilson denken.
→ Therapie: Therapieziel ist initial eine negative - im weiteren Behandlungsverlauf ausgeglichene Kupferbilanz. Hierfür stehen Chelatbildner (führen zur Entspeicherung von Kupfer aus der Leber) und Zinksalze (hemmen die Kupferaufnahme aus dem Darm) bereit.
→ I: Allgemein:
→ 1) Kupferarme Diät; Meiden von Meeresfrüchten, Innereien, Kakao etc.
→ 2) Die Applikation von Zinksulfat, 2 x 75 mg/d zwischen den Mahlzeiten hemmt durch gesteigerte Synthese von Metallothionein in den Mucosazellen des Darms die enterale Kupferresorption.
→ 3) Die Applikation von Vitamin B6 zur Prophylaxe einer Optikusneuropathie.
→ II: Medikamentöse Therapie: Ist insbesondere bei symptomatischen Patienten indiziert und sollte für mindestens 6 Monate (bis 1 Jahr) durchgeführt werde:
→ 1) Trientine /Triethylentetramin: 1,2-2,7g/d 3 x vor den Mahlzeiten: Ist das Mittel der ersten Wahl, aufgrund der wenigen Nebenwirkungen.
→ 2) D-Penicillamin: Einschleichende Dosierung bis zu einer mittleren Tagesdosis von 900-1800mg/d vor den Mahlzeiten. Hierbei handelt es sich um einen Chelatbildner, der die Ausscheidung von Kupfer fördert. Nebenwirkungen sind insbesondere Hautausschlag, Fieber, Leuko-, Thrombozytopenie, nephrotisches Syndrom, SLE, Goodpasture-Syndrom und nicht zuletzt die Myasthenie. Aufgrund der ausgeprägten Nephrotoxizität sollten die Nierenwerte engmaschig kontrolliert werden.

→ III: Lebertransplantation: Die Lebertransplantation wird insbesondere bei der fulminater Form durchgeführt; sie stellt die einzige kurative Therapieoption beim Morbus Wilson dar.
→ Prognose: Bei früh einsetzender Therapie und guter Compliance des Patienten mit engmaschigen Therapie- und Verlaufskontrollen exzellent; der Spontanverlauf des Morbus Wilson ist immer letal endend.