→ Definition: Bei der paroxysmalen nächtlichen Hämoglobinurie handelt es sich um die einzig erworbene korpuskuläre hämolytische Anämie; sie beruht auf einem klonalen Defekt der hämatopoetischen Stammzellen.
→ Epidemiologie: Die paroxysmale nächtliche Hämoglobinurie stellt mit einer Inzidenz von 1: 1000000 Einwohner eine sehr seltene Erkrankung dar und hat einen Manifestationsgipfel zwischen dem 25.-45. Lebensjahr, wobei keine Geschlechterprävalenz existiert.
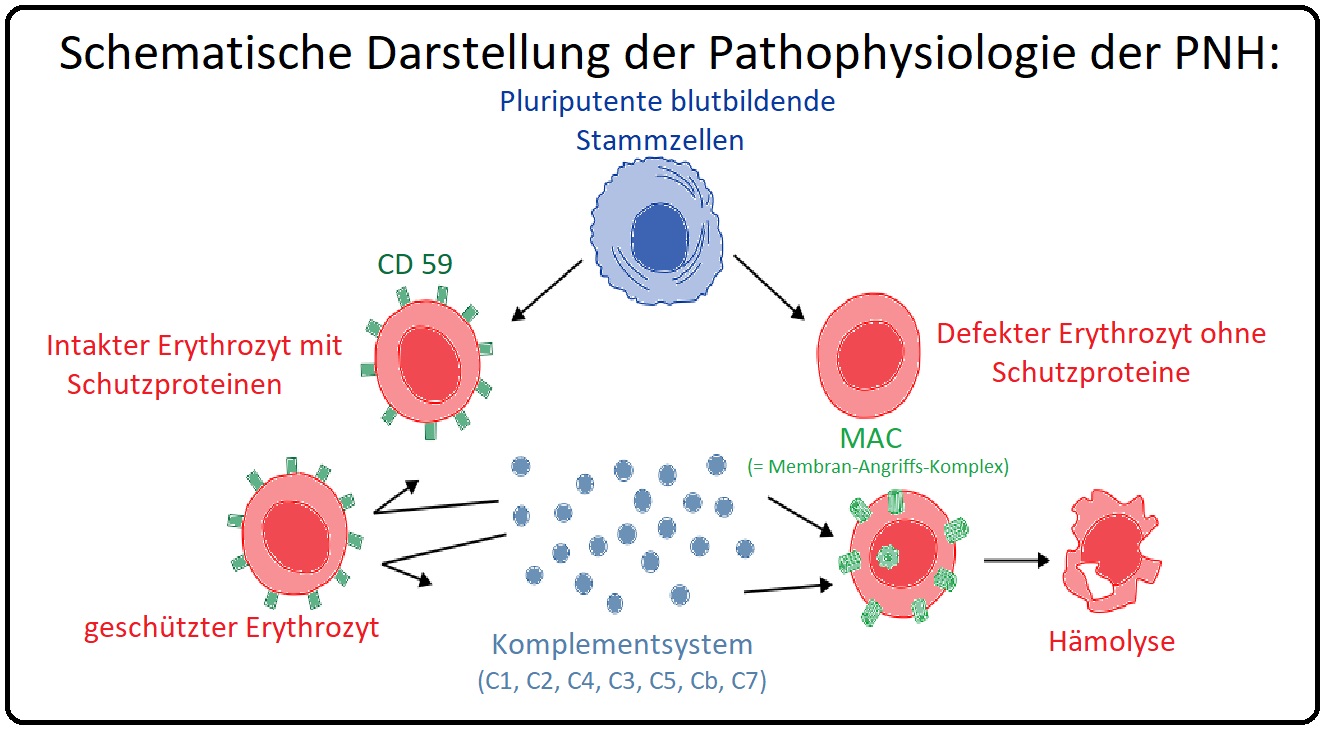
→ Ätiopathogenese: Im Mittelpunkt der Erkrankung steht eine erworbene somatische Spontanmutation im X-chromosomal kodierten PIG-A-Gen mit konsekutiver defekter Synthese des Glykosylphosphatidylinosityl-Ankers (GIP-Anker) in ein oder mehreren pluripotenten Stammzellen.
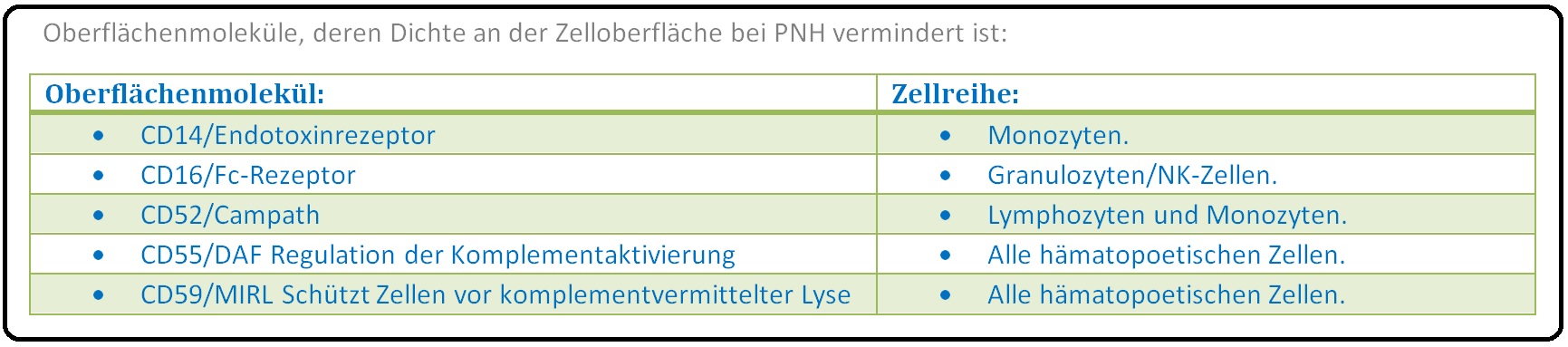
→ I: Der GIP-Anker hat die Aufgabe verschiedene funktionell relevante Oberflächenproteine in der Zellmembran zu verankern.
→ II: Somit ist auch die Dichte verschiedener Proteine wie z.B. komplementinaktiverende Faktoren DAF (= Decay-Accelerating-Factor) und MIRL (= Membrane-Inhibitor of reactive-Lysis) vermindert. Die Folge ist eine vermehrte komplementvermittelte intravasale Lyse der Erythrozyten.

→ III: Die Schwere der Hämolyse hängt insbesondere vom Anteil der abnormen Zellen und dem Grad der Komplementaktivierung ab. Die Komplementaktivierung ist besonders ausgeprägt, wenn eine Azidose besteht z.B. nachts (bei möglichen Apnoephasen), im Rahmen von Infektionen, Traumen oder Operationen.
→ IV: Charakteristischerweise weist die Hämolyse einen schubweisen oder chronischen Krankheitsverlauf auf und führt zur Hämoglobinurie, die mit einer schwärzlichen Verfärbung des Morgenurins einhergeht.
→ V: Da sich der Defekt schon auf der Ebene der pluripotenten Stammzellen manifestiert, sind nicht selten alle hämopoetischen Zellreihen betroffen; so zeigt sich bei 30% der Betroffenen gleichzeitig eine Hypoplasie der Thrombo- und Granulozytopoese.
→ Klinik: Der Krankheitsverlauf der paroxysmalen nächtlichen Hämoglobinurie ist sehr wechselhaft und es zeigt sich zumeist eine schubweise Progredienz, aber auch Spontanremissionen sind möglich.
→ I: Allgemeinsymptome mit Schwäche und Leistungsminderung (= Fatigue) aufgrund der intravasalen Hämolyse.
→ II: Die paroxysmale nächtliche Hämoglobinurie manifestiert sich klassischerweise nach dem Schlaf, aber auch nach körperlicher Anstrengung, Fieber, Gabe von Kontrastmittel, etc. (es fällt so viel Hämoglobin an, dass es nicht durch Haptoglobin gebunden werden kann).
→ III: Hämolytische Krisen: Mit Schmerzattacken. Die Patienten klagen über Bauch-, Rücken- und Kopfschmerzen sowie Dyspnoe infolge von Durchblutungsstörungen.
→ IV: Weitere Symptome: Sind insbesondere:
→ 1) Nierenfunktionsstörungen,
→ 2) Eisenmangel bedingt durch den renalen Eisenverlust bei Hämoglobinurie.
→ 3) Thromboembolische Ereignisse treten sowohl an typischen (venös) als auch atypischen Lokalisationen (arteriell, in Lebervenen, Pfortader, etc.); wichtige thromboembolische Ereignisse sind u.a. die tiefe Beinvenenthrombose, Lungenembolie, TIA, Stroke, Myokardinafarkt, Pfortaderthrombose, Budd-Chiari-Syndrom, etc.
→ 4) Nicht selten manifestiert sich eine mäßige Splenomegalie und ein leichter intermittierender Ikterus.
→ Klinisch-relevant: Die Ursachen der erhöhten Thromboseneigung ist komplex; zwei wichtige Annahmen hierbei sind zum einen die NO-Depletion durch Hämoglobin, als auch eine vermehrte Expression von Tissue-Faktor durch Komplementaktivierung.
→ Komplikationen: Wichtige und z.T. schwerwiegende Komplikationen sind bei der PNH u.a.:
→ I: Schwerwiegende thromboembolische Ereignisse mit atypischer Lokalisation (Pfortader-, Sinusvenenthrombose, etc.) mit z.T. hoher Mortalität.
→ II: Spätkomplikationen sind insbesondere die Myelodysplasie sowie die akut myeloische Leukämie.
→ Diagnose: Im Vordergrund der klinischen Diagnostik steht die Laborchemie:
→ I: Anamnese/klinische Untersuchung: Mit Eruierung von Anämiesymptomen, Leistungsminderung, Schwäche, rezidivierende Abdominalschmerzattacken, Dysphagie, eines dunkel gefärbten Morgenurins, etc.
→ II: Labor:
→ 1) Nachweis einer Anämie (normo-, makrozytär bei Retikulozytose, mikrozytär bei Eisenverlust) mit positiven Hämalysezeichen (HB, LDH, Bilirubin und Retikulozyten erhöht, HK und Haptoglobin erniedrigt). Insbesondere in der Phase der hämolytischen Krise ist die Retikulozytenzahl deutlich erhöht (10- 20%; Norm 0,5-1,5%).
→ 2) Möglicher Nachweis einer Thrombo- und Leukozytopenie (Panzytopenie) als Ausdruck einer hämatopoetischen Bildungsstörung.
→ 3) In der Urinanalyse lässt sich eine Hämoglobinurie (dunkler Morgenurin) und eine Hämosiderinurie darstellen.
→ Klinisch-relevant: Der Coombs-Test ist charakteristischerweise negativ, sodass bei jeder Coombs-negativen Hämolyse immer an eine paroxysmale nächtliche Hämoglobinurie gedacht werden muss. Ebenso auch bei Panzytopenie mit Retikulozytose.
→ III: Die Diagnosesicherung erfolgt durch den direkten Nachweis (PIG-A-Mutation) mittels PCR oder des Membrandefektes mittels durchflusszytometrischer Bestimmung der GPI-verankerten-Antigene (CD55 und CD59) auf Erythrozyten und Granulozyten.
→ IV: Bildgebung: Oberbauchsonographie einschließlich der Doppleruntersuchung mit Bestimmung von:
→ 1) Milz- und Lebergröße.
→ 2) Dopplersonographisch möglicher Nachweis einer Lebervenen-, Pfortader-, Milzvenen oder Mesenterialvenenthrombose (abgelaufen oder akut).
→ Differenzialdiagnose: Von der paroxysmalen nächtlichen Hämoglobinurie müssen insbesondere nachfolgende Erkrankung abgegrenzt werden:
→ I: Myelodysplastische und myeloproliferative Syndrome.
→ II: Thrombotische-thrombozytopenische Purpura (mit Nachweis von Fragmentozyten im Blutausstrich).
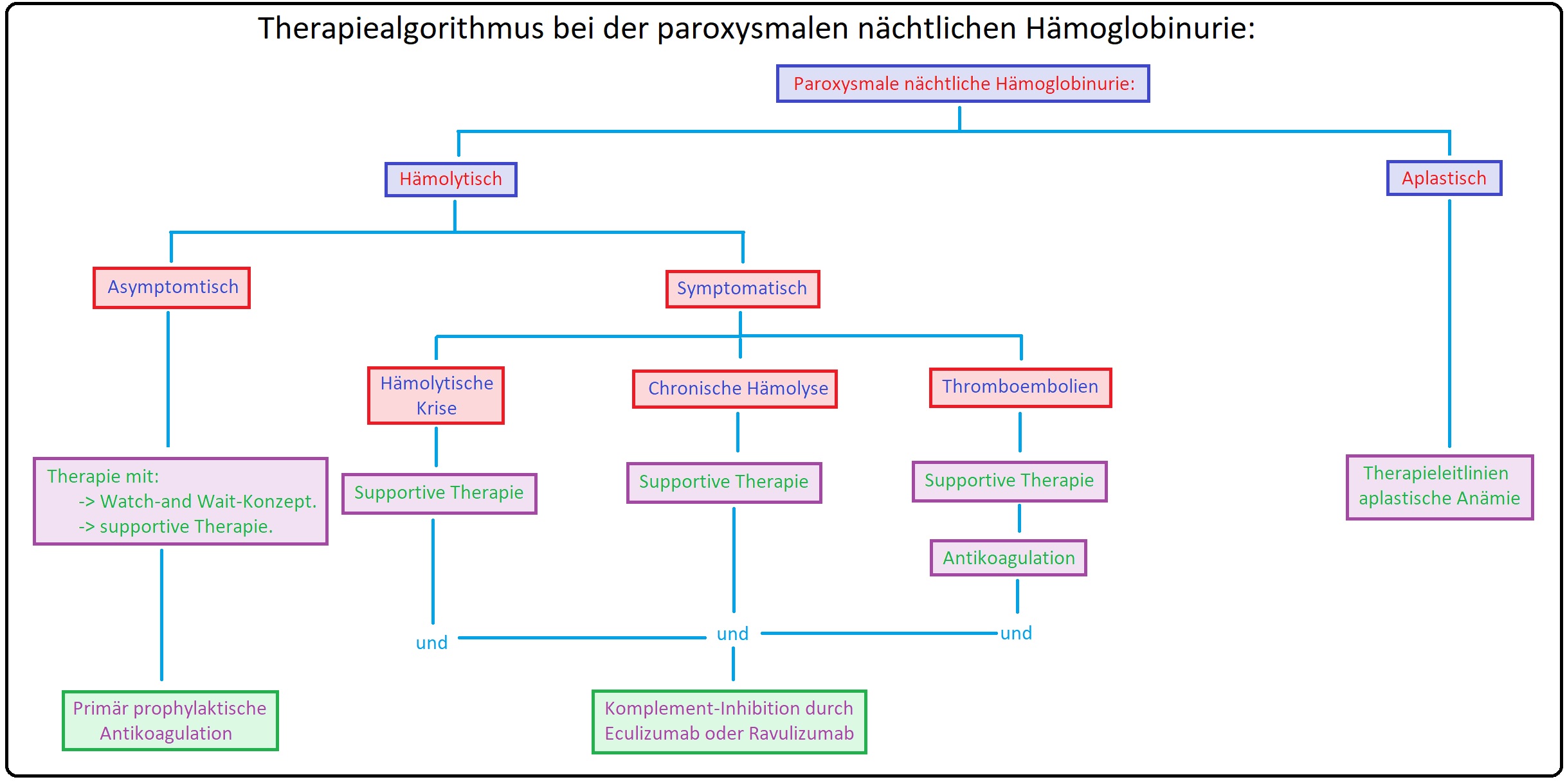
→ Therapie: Zeigt sich ein asymptomatischer Krankheitsverlauf kann ein „Watch-and-Wait“ Therapiekonzept in Betracht gezogen
werden.
→ I: Supportive Therapie:
→ 1) Bei symptomatischer Anämie ist die Applikation eines AB0-kompatiblen Erythrozytenkonzentrates indiziert. Zusätzlich kann Folsäure (1-5mg/d) oder Vitamin B12 bei massiv gesteigerter Erythropoese im Knochenmark verabreicht werden.
→ 2) Evtl. orale Substitution von Eisen (unter Kontrolle der Eisenspeicher Ferritin, Transferrin-Sättigung, etc.); eine intravenöse Applikation sollte vermieden werden, da ein hämolytischer Schub ausgelöst werden kann.
→ 3) Infektionen sollten frühzeitig und adäquat behandelt werden, um eine Exazerbation der paroxysmalen nächtlichen Hämoglobinurie zu vermeiden.
→ II: Des Weiteren ist nach stattgefundener Thrombose eine lebenslange Antikoagulation indiziert. Insbesondere bei großem PNH-Klon (> 50% der Granulozyten) kann eine primär prophylaktische Antikoagulation erwogen werden.
→ III: Komplement-Inhibition: Seit 2007 steht ein humanisierter monoklonaler Antikörper, Eculizumab bzw. Ravulizumab, ein Inhibitor des terminalen Komplementsystems zur Verfügung. Sie binden an den Komplementfaktor C5, verhindert dessen Spaltung in die Fragmente C5a und C5b und hemmt somit die Bildung des terminalen Komplementkomplexes C5b-9. Die Dosierung erfolgt initial mit 600mg intravenös 1x/Woche für 4 Wochen und anschließend mit einer Erhaltungsdosierung von 900mg alle 2 Wochen. Ziel hierbei ist die Verbesserung der hämolytischen Symptome. Indikationen sind u.a.:
→ 1) Patienten mit hohem Hämolyse-bedingten Transfusionsbedarf.
→ 2) Nach thromboembolischen Ereignissen.
→ 3) Bei PNH-assoziierter Niereninsuffzienz,
→ 4) Abdominelle Schmerzkrisen und weitere Symptome.
→ Klinisch-relevant: Unter Eculizumab kommt es durch Blockade der terminalen Komplementstrecke zu einem erhöhten Risiko für Infektionen mit kapselbildenden Bakterien wie die Meningokokken. Diesbezüglich sollte vor jedem Therapiebeginn eine Meningokokken-Schutzimpfung durchgeführt werden.
→ IV: Allogene Stammzellentransplantation: Stellt die einzige kurative Therapie dar und ist insbesondere bei jüngeren Patienten mit schweren Krankheitsverläufen (rezidivierende aplastische Krisen), Entwicklung einer AML oder eines dysplastischen Syndroms indiziert.
→ Prognose: Die Lebenserwartung (bzw. mediane Überlebenszeit) ist bei der paroxysmalen nächtlichen Hämoglobinurie verkürzt und beträgt etwa 22 Jahre.
→ I: Todesursachen sind insbesondere thromboembolische Ereignisse sowie das Budd-Chiari-Syndrom.
→ II: Auch werden Übergänge in das myelodysplastische Syndrom oder in eine akute Leukämie beobachtet.