- Details
- Geschrieben von: CF
- Kategorie: Phakomatosen
- Zugriffe: 3329
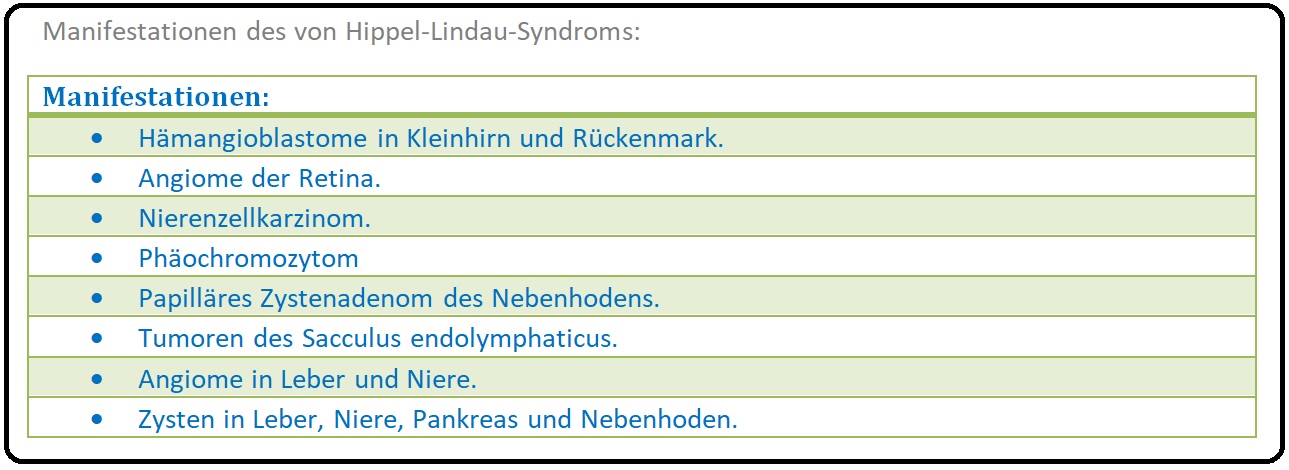
→ Definition: Bei der von Hippel-Lindau-Krankheit handelt es sich um eine autosomal-dominant vererbte mesenchymale Dysplasie, die durch das Auftreten von:
→ I: Angiomatosis retinae (von Hippel),
→ II: Das Hämangioblastom des Kleinhirns (= Lindau-Tumor) oder ein spinales Hämangioblastom sowie durch
→ III: Viszeral zystische Veränderungen gekennzeichnet ist. Sie wird neben der Neurofibromatose, tuberösen Sklerose, dem Sturge-Weber-Syndrom, etc. zu den Phakomatosen gezählt.

→ Epidemiologie:
→ I: Das von Hippel-Lindau-Syndrom weist eine Inzidenz von 1/36000 und beruht auf einer Keimbahnmutation auf dem kurzen Arm von Chromosom 3 (3p25/p26).
→ II: Der Manifestationgipfel liegt zwischen dem 18.-50. Lebensjahr (tritt zumeist vor dem 30. Lebensjahr auf), wobei beide Geschlechter gleichermaßen betroffen sind.
→ Genetik:
→ I: Das von Hippel-Lindau-Gen besitzt 3 Exons und kodiert ein aus 213 Aminosäuren bestehendes nukleäres Protein (= pVHL).
→ II: pVHL-Funktion:
→ 1) pVHL spielt eine wichtige Rolle beim ubiquitinabhängigen proteasomalen Abbau von HIF-1-Alpha (= hypoxia-inducible-faktor).
→ 2) HIF-1-Alpha reguliert wiederum die Transkription verschiedener hypoxieinduzierbarer Gene für z.B. den vaskulären endothelialen Wachstumsfaktor (VEGF), den Plättchenwachstumsfaktor (PDGFB), den transformierenden Wachstumsfaktor-Alpha (TGFA) und Erythropoetin.
→ 3) Eine Inaktivierung des pVHL induziert somit eine verstärkte Expression HIF-1-Alpha regulierter Zielgene, was letztendlich zur Tumorproliferation, Neovaskularisation und Bildung von Erythropoetin führt. Zudem hat pVHL Einfluss auf die Tumorgenese (die Mechanismen sind bis jetzt nicht genau bekannt).

→ Klinik: Die Symptomatik der von Hippel-Lindau-Krankheit ist sehr variabel und insbesondere von der Lokalisation des Tumors abhängig.
→ I: Das Krankheitsbild kann insbesondere zu Beginn klinisch stumm sein, bis plötzlich Einklemmungssymptome mit unerträglichen okzipitalen Kopfschmerzen auftreten. Es können sich aber auch Visusstörungen, Kopf- oder Rückenschmerzen sowie ein geringer Nystagmus zu Krankheitsbeginn manifestieren.
→ II: ZNS:
→ 1) Kopfschmerzen im Hinterkopfbereich,
→ 2) Im weiteren Krankheitsverlauf Hirndruckzeichen mit Übelkeit, Erbrechen, Okulomotorikstörung, Schluckstörungen Ataxie, bei tiefsitzenden Tumoren progrediente Querschnittslähmung durch Spinalkompression, etc.
→ III: Augen: Das retinale Angiom kann zu Sehstörungen (z.B. Visusminderung, etc.), Netzhautablösung und letztlich zur Erblindung führen.
→ IV: Lindau-Zysten: (= Hämangioblastome) Insbesondere im Bereich der Nieren und des Pankreas, aber auch in Leber, Lunge oder Nebenhoden.
→ V: Komplikationen: Das von Hippel-Lindau-Syndroms ist vermehrt mit nachfolgenden Krankheitsbildern vergesellschaftet:
→ 1) Nierenzellkarzinom (häufig bilateral oder multifokal),
→ 2) Phäochromozytom mit Kopfschmerzen, Schwindel, Herzrasen und hypertensiven Krisen.
→ 3) Polyzythämie (aufgrund einervermehrten Erythropoetin-Produktion durch die Angiome).
→ Diagnose: Im Vordergrund der Diagnose steht die radiologische Bildgebung.
→ I: CT/MRT:
→ 1) Das Hämangioblastom stellt sich als scharf begrenzte, homogene, flüssigkeitsgefüllte Zyste dar, in der sich ein kleiner gut vaskularisierter Tumorknoten mit deutlicher Kontrastmittelanreicherung befindet.
→ 2) Möglicher computertomographischer Nachweis von Nieren- und Pankreaszysten sowie des Nierenzellkarzinoms.
→ II: Angiographie: Spielt insbesondere in der präoperativen Planung eine wichtige Rolle mit Darstellung eines kräftig kontrastierten Gefäßkonvoluts in der frühen arteriellen Phase.
→ III: Laborchemische Untersuchung:
→ 1) Nachweis einer Polyglobulie aufgrund einer gesteigerten Sekretion von Erythropoetin (durch den Tumor).
→ 2) Bestimmung der Katecholamine im 24h-Urin (Phäochromozytom).
→ IV: Ophthalmologische Untersuchung in Mydriasis mit Nachweis von Hämangioblastomen in der Retina und evtl. Glaukom-Nachweis.

→ Differenzialdiagnose: Von der von Hippel-Lindau-Krankheit müssen u.a. nachfolgende Erkrankungen abgegrenzt werden:
→ I: Weitere Phakomatosen wie die tuberöse Sklerose, das Sturge-Weber-Syndrom, etc.
→ II: Isolierte zerebelläre Hämangioblastome und AV-Angiome.
→ III: Des Weiteren zerebrale Metastasen oder das pilozytische Astrozytom.
→ Therapie: Eine evidenzbasierte kausale Behandlung existiert für das von Hippel-Lindau-Syndrom bis heute nicht.
→ I: Operative Therapie:
→ 1) Mittel der Wahl stellt die vollständige Exstirpation des Tumors (Zyste und pialer muraler Knoten) dar, da ansonsten die Rezidivrate deutlich erhöht ist. Präoperativ ist die angiographische Darstellung der vaskulären Versorgung obligat.
→ 2) Bei großen Tumoren kann eine präoperative Devaskularisation durch Embolisation eine Verkleinerung und dadurch die Operabilität ermöglichen.
→ II: Ophthalmologie: Retinale Angiome werden mittels Laserkoagulation therapiert.
→ Prognose: Die Prognose der von Hippel-Lindau-Krankheit wird insbesondere durch die extrazerebral malignen Tumorerkrankungen bestimmt (metastasierendes Nierenzellkarzinom mit hoher Letalität), sodass regelmäßige Kontrolluntersuchungen auf Nierenzellkarzinom und Phäochromozytom nötig sind. Auch wird durch die Applikation einer antiangiogenen Substanz versucht, das Tumorwachstum zu beeinflussen.
- Details
- Geschrieben von: CF
- Kategorie: Phakomatosen
- Zugriffe: 5140
→ Definition: Beim Sturge-Weber-Syndrom handelt es sich um eine angeborene enzephalotrigeminale Angiomatose aus der Gruppen der Phakomatosen (= neurokutane Krankheiten mit Hämangiombildung im Gesicht, an den Meningen und der Chorioidea), zu denen auch die von- Hipple-Lindau-Krankheit, die Neurofibromatose und die tuberöse Sklerose zählen. Klinische Charakteristika dieses Syndroms sind u.a.:
→ I: Naevus flammeus im Gesicht,
→ II: Ipsilaterale Hämangiome der Meningen und einer damit verbundenen reaktiven Hirnatrophie.
→ Epidemiologie:
→ I: Das Sturge-Weber-Syndrom stellt mit einer Prävalenz von 0,5/100000 eine sehr selten Erkrankung dar, wobei beide Geschlechter gleichermaßen betroffen sind.
→ II: Die Erkrankung tritt zumeist spontan auf und wird nicht monogen vererbt; jedoch existiert eine familiäre Häufung.
→ Ätiopathogenese: Im Mittelpunkt der Genese des Struge-Weber-Syndroms steht die mangelnde Differenzierung des embryonalen Gefäßplexus (zwischen dem 2.-4. Schwangerschaftsmonat) mit konsekutivem Verbleiben von dünnwandig erweiterten Kapillaren und Venen. Sie sind in der Regel an der Gesichtshaut, der Leptomeninx (= Bindegewebsschicht der Meningen, die einen geringen Faseranteil aufweist und so als "weiche Häute" des ZNS bezeichnet wird) des gleichseitigen Okzipitallappens und der Corioidea des Auges nachweisbar. Durch eine pathologisch venöse Stase und insuffiziente Drainage kommt es in der Folge zu eine fokalen Atrophie der Retina und einer Gliose mit konsekutiver Verkalkung der an die Leptomenix angrenzenden Kortex. In ausgeprägten Fällen manifestiert sich eine Hemiatrophie des Gehirns.
→ Klinik: Bei voller klinischer Ausprägung des Sturge-Weber-Syndroms besteht eine typische Symptomtrias (die zumeist nur sehr selten anzutreffen ist):
→ I: Haut:
→ 1) Naevus flammeus (= kutanes Hämangiom) oder Portwein-Naevus einseitig insbesondere im Versorgungsbereich des 1. Trigeminusastes.
→ 2) Weitere Auffälligkeiten sind eine allgemeine Gesichtsasymmetrie, eine ipsilaterale Visusminderung sowie ein Buphthalmus (= Bulbusvergrößerung).
→ II: Neurologische Symptome:
→ 1) Mentale Retardierung und fokale oder generalisierte epileptische Anfälle (v.a. komplexe Partialanfälle).
→ 2) Evtl. Entwicklung einer allmählichen oder schubweisen Hemianopsie und Hemiparese.
→ III: Kongenitales Glaukom.
→ IV: Weitere Symptome: Sind u.a.:
→ 1) Kopfschmerzen, die den Charakter einer Migräne haben.
→ 2) Störungen der Persönlichkeitsentwicklung, im weiteren Krankheitsverlauf können sich dann Wesenveränderungen und/ oder eine Demenz manifestieren.
→ Diagnose: Im Mittelpunkt der Diagnostik stehen die bildgebenden Verfahren (CT und MRT).
→ I: Anamnese/klinische Untersuchung: Eruierung einer Entwicklungsstörung, inspektorischer Nachweis eines Naevus flammeus im Versorgungsgebiet v.a. des 1. Trigeminusastes oder auch der anderen Äste, etc. 1/3 der Pattienten weisen schon im Wachstumsalter eine Hemiparese mit Hypotrophie der betroffenen Extremität und/oder eine homonyme Hemianopsie auf.

→ II: Bildgebende Verfahren:
→ 1) Im CT zeigen sich im okzipitalen Bereich kortikale und subkortikale Verkalkungen in Form von die Gyri und Sulci nachzeichnende girlandenförmige Verschattungen.
→ 2) Kernspintomographisch sind flächige Verdickungen der Pia mater mit ausgeprägter Kontrastmittel-Aufnahme entsprechend eines meningealen Angioms eruierbar. Weitere Auffälligkeiten des Sturge-Weber-Syndroms sind insbesondere:
→ A) Volumenminderung der betroffenen Hemisphäre mit Verlagerung der Mittellinienstrukturen.
→ B) Verdickung der Schädelkalotte, Schädelasymmetrie sowie Vergrößerung des ipsilateralen Sinus frontalis.
→ C) Vermehrte KM-Aufnahme im Plexus coroideus auf der gleichen Hemisphäre wie das piale Angiom.
→ III: EEG: Mit möglichem Nachweis einer Anfallsbereitschaft und/oder Herdbefunden.
→ IV: Ophtalmologische Untersuchung: Hierbei sind Ausschluss nachfolgender Erkrankungen (z.B. Chorioidea-Angiom, Hemianopsie, Netzhautablösung, Glaukom, etc.) obligat.
→ Differenzialdiagnose: Vom Sturge-Weber-Syndrom müssen insbesondere nachfolgende Erkrankung abgegrenzt werden:
→ I: Weitere Phakomatosen wie die von-Hippel-Lindau-Krankheit, Neurofibromatose, etc.
→ II: Isolierte arteriovenöse Angiome, die jedoch weniger zur Verkalkung neigen und tief in das Hirnparenchym hereinreichen.
→ Therapie: Eine kausale Therapie existiert beim Sturge-Weber-Syndrom nicht, vielmehr steht die konsequente antiepileptische Therapie im Vordergrund.
→ I: Lasertherapie: Kutaner Hämangiome sowie eine photodynamische Therapie mit Verteporfin bei bestehendem Chorioidea-Hämangiom.
→ II: Eine chirurigische Intervention kann bei therapierefraktären Fällen (sofern das Angiom einseitig ist und vollständig entfernt werden kann) erwogen werden.
→ Prognose: Die Prognose ist insbesondere vom Ausmaß der Minderperfusion der angrenzenden Kortex abhängig. Je früher sich vaskuläre Komplikationen manifestieren, umso eher wird die intellektuelle und motorische Entwicklung beeinträchtigt.