→ Definition: Das Phäochromozytom ist definiert als ein katecholamin-produzierender, von chromaffinen Geweben ausgehender, zumeist benigner neuroendokriner Tumor des Nebennierenmarks oder der Paraganglien des Grenzstrangs (sympathisches Ganglion des Brustraums = Paragangliom). Dieser Tumor produziert und sezerniert unkontrolliert Katecholamine (Katecholamine werden aus der Aminosäure L-Tyrosin synthetisiert über L-Tyrosin → L-Dopa → Dopamin → Noradrenalin → Adrenalin), die zur krisenhaften oder auch dauerhaften arteriellen Hypertonie führen.

→ Lokalisation:
→ I: 85% der neuroendokrinen Tumoren sind gutartig und im Bereich des Nebennierenmarks (NNM) lokalisiert; sie produzieren Noradrenalin mehr als Adrenalin.
→ II: 10%-(15%) sind bösartig und bilden Noradrenalin und Dopamin; Risikofaktoren hierfür sind insbesondere junges Alter, weibliches Geschlecht, Tumorgröße > 5cm sowie extraadrenale Manifestation.
→ III: 90% treten einseitig (solitär) auf, 10% haben eine beidseitige Lokalisation (insbesondere bei der familiären Form).
→ IV: Extraadrenale Phäochromozytome (= Paragangliome) können überall entstehen, wo sympathisches Nervengewebe vorhanden ist. Das größte extraadrenale Paraganglion ist das Zuckerkandl-Organ, das vor der distalen Aorta oder in Höhe der Aortenbifurkation gelegen ist.

→ Epidemiologie:
→ I: Das Phäochromozytom stellt mit einer Inzidenz von 1/100000/Jahr eine seltene Erkrankung dar; es ist in 0,1-0,2% der Fälle Ursache einer arteriellen Hypertonie.
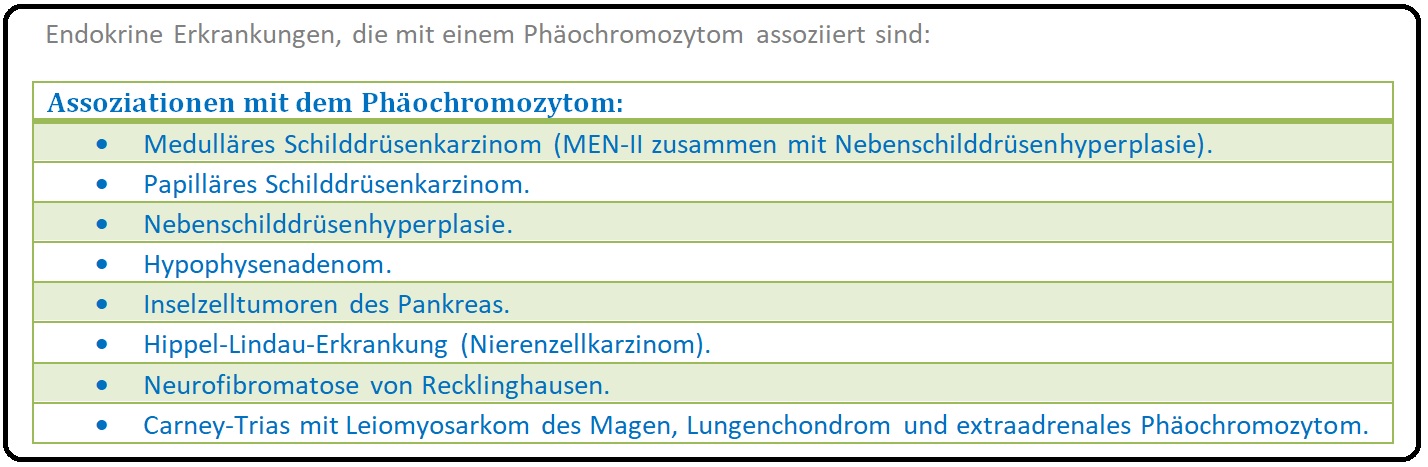
→ II: Der Manifestationsgipfel liegt zwischen dem jüngeren und mittleren Lebensalter (40.-50. Lebensjahr), wobei Männer und Frauen gleichermaßen betroffen sind; die hereditiäre Formen (z.B. bei MEN II, Neurofibromatose von Recklinghausen, von Hippel-Lindau-Erkrankung, etc.) findet man < 40. Lebensjahr selten sogar im Kindesalter (10% der Phäochromozytome werden im Kindesalter diagnostiziert).
→ Ätiologie:
→ I: Ursache nicht genau bekannt.
→ II: In 25% der Fälle findet man eine hereditäre Genese:
→ 1) MEN Typ 2a: Die multiple endokrine Neoplasie (= Sipple-Syndrom) Typ 2a mit medullärem Schilddrüsen-Karzinom, Hyperparathyreoidismus, Phäochromozytom und Neurinomen wird durch eine Mutation des RET-Protoonkogens verursacht. Das Phäochromozytom tritt hierbei häufiger (im Vergleich zur sporadischen Form) bilateral und multifokal auf.
→ 2) Von Hippel-Lindau-Syndrom: (= von-Hippel-Lindau-Krankheit) Es besteht eine Mutation des VHL-Gens mit Ausbildung eines neurokutanen Syndroms. Das klinische Bild ist geprägt durch Gefäßmissbildungen (retinale Hämangiomatose, bzw. zerebrale Hämangiomatose), das klarzellige Nierenzellkarzinom und das Phäochromozytom.
→ 3) Neurofibromatose Typ 1: Mit Mutation des Neurofibromatose-Gens (Morbus Recklinghausen). Hierdurch entwickelt sich ein Mangel an Neurofibrin, einem Regulatorprotein. Die Klinik ist durch das Auftreten multipler Neurofibrome der Haut, aber auch des GIT und der Nerven, Cafe-au-lait-Flecken, Lisch-Knoten (= auf der Iris befindliche pigmentierte Hamartome) gekennzeichnet. Weiterhin besteht eine erhöhtes Risiko für Meningiome, Gliome, und das Phäochromozytom.
→ Pathophysiologie: Im Nebennierenmark existieren 2 katecholaminproduzierende Zelltypen, die Adrenalin- und Noradrenalinzellen. Vorläufer dieser biogenen Amine ist das Tyrosin, das durch die Tyrosinhydroxylase in Dopa und anschließend in Dopamin sowie Noradrenalin metabolisiert wird; beide Amine werden in der intrazellulären Granula gespeichert. Nur im Nebennierenmark kann Noradrenalin mit Hilfe der Phenylethanolamin-N-Methyltransferase in Adrenalin methyliert werden (extraadrenale Tumoren können kein Adrenalin, jedoch Noradrenalin und Dopamin synthetisieren).
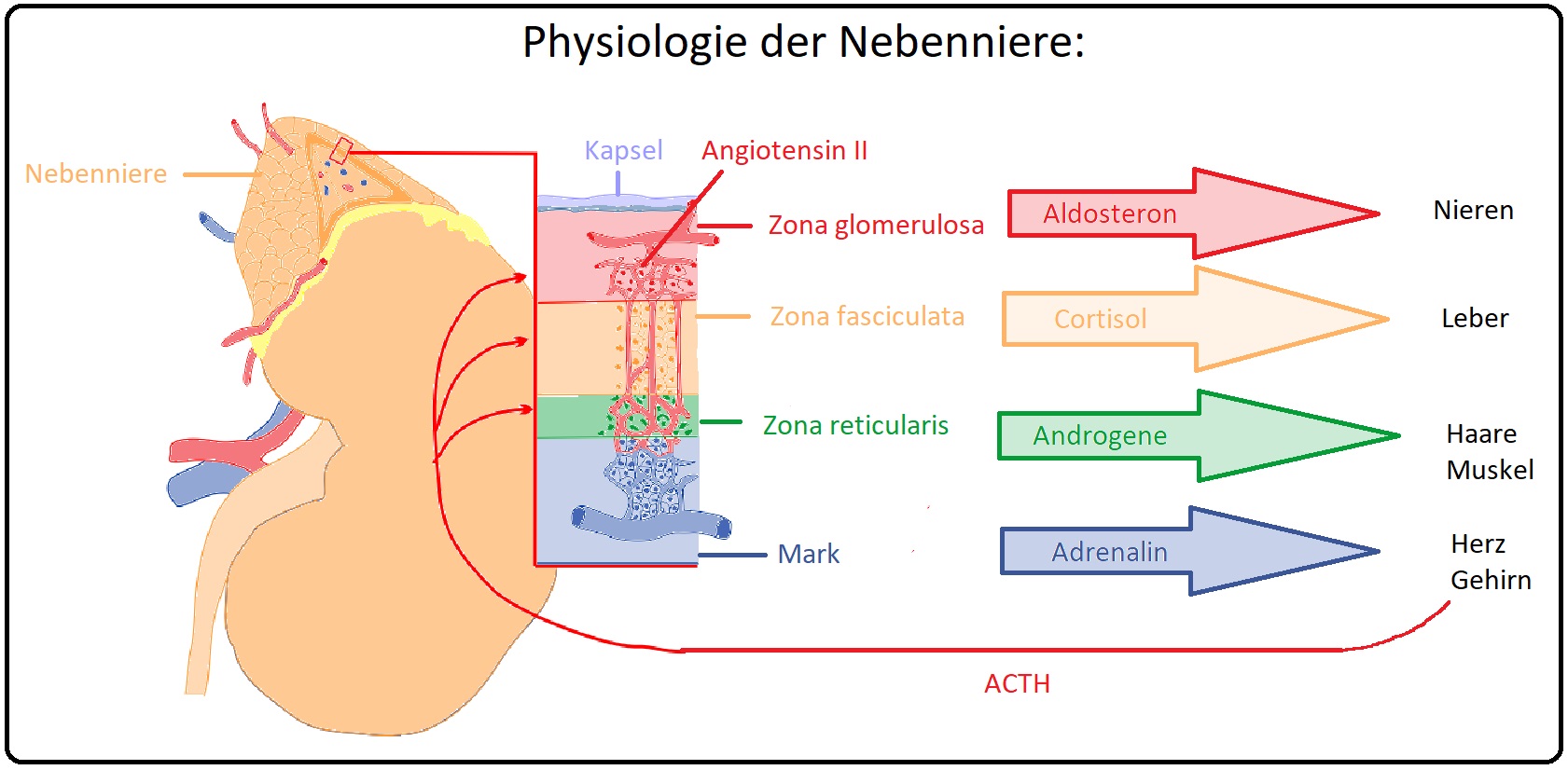
→ Klinik:
→ I: Die Kardinalssymptome des Phäochromozytoms sind insbesondere:
→ 1) Anfallsweise Kopfschmerzen (pulssynchron).
→ 2) Hyperhidrosis.
→ 3) Tachykardie mit Herzrasen und Palpitation. Triggermechanismen sind insbesondere psychische Stressoren, Angst, Nikotin, etc.
→ II: Weitere Leitsymptome sind paroxysmale arterielle Hypertonie, persistierende arterielle Hypertonie sowie hypertensive Krisen/Notfälle.
→ III: Sehstörungen (und Augenhintergrundsveränderungen), Schwindel, Schwäche, Ohrensausen, selten Tremor, innerer Unruhe, Übelkeit, Erbrechen, Angst, etc.
→ IV: Weitere Symptome: Sind Hautblässe/Gesichtsblässe aufgrund peripherer Vasokonstriktion, Hyperglykämie mit Glucosurie, Gewichtsverlust (aufgrund eines Hypermetabolismus), aber auch Fieber und Leukozytose.
→ Klinisch-relevant:
→ A) Häufig manifestieren sich frühzeitig Endorganschäden wie hypertensive Retinopathie, linksventrikuläre Hypertrophie, Nephrokalzinose etc. aufgrund der ausgeprägten arteriellen Hypertonie; zudem ist das Phäochromozytom nicht selten mit einer Tako-Tsubo-Kardiomyopathie vergesellschaftet.
→ B) Bei Patienten, die eine Gewichtszunahme und Gesichtsröte aufweisen, kann ein Phäochromozytom mit hoher Wahrscheinlichkeit ausgeschlossen werden.
→ C) Nach Gabe von ß-Blockern kann sich ein paradoxer Blutdruckanstieg manifestieren.
→ Komplikationen: Insbesondere bei krisenhaften klinischen Krankheitsverläufen können sich lebensbedrohliche Komplikationen entwickeln. Hierzu zählen:

→ Diagnose:
→ I: Anamnese: Eruierung einer schweren und medikamentös schlecht einstellbaren Hypertonie sowie möglicher Triggermechanismen wie physischer wie psychischer Stress, Lagewechsel, Druck auf die Tumorgegend (Husten, Pressen, Palpation), Gabe von Beta-Blockern, etc. Klinisches Bild einer hypertensiven Krise mit Kopfschmerzen, Schwitzen, Herzklopfen, Hautblässe und fehlender Blutdruck- Nachtabsenkung ist immer Phäochromozytom-verdächtig.
→ Klinisch-relevant: Phäochromozytom-verdächtig sind auch immer:
→ A) Neu auftretende therapieresistente Hypertonie,
→ B) Akut auftretende hypertensive Krise während der Narkose bzw. eines operativen Eingriffs.
→ II: Labor: Allgemein sollte darauf geachtet werden, dass interferierende Medikamente wie z.B. Tetrazykline, Clonidin oder Theophyllin, etc. 2 Wochen vor der Labordiagnostik abgesetzt werden; zudem muss vor der Blutentnahme eine 30-minütige Ruhelagerung des Patienten erfolgen.
→ 1) Bestimmung der Katecholaminmetaboliten Metanephrin (112ng/l = 0,6mmol/l normal mit hoher Sensitivität und Spezifität) und Normetanephrin (61ng/l = 0,3mmol/l normal) im Plasma. Werte die > 3fache erhöht sind deuten auf ein Phäochromozytom hin. Evtl. Nachweis einer Hyperglykämie, Leukozytose oder einer Laktatazidose.
→ 2) Besteht ein Phäochromozytom ist zur Abklärung einer MEN-2 immer auch die Kalzium- und Calcitonin-Bestimmung obligat.
→ III: Urin:
→ 1) Bestimmung der Katecholamine (Adrenalin, Noradrenalin) bzw. Katecholaminmetaboliten im angesäuerten 24-Stunden-Urin; Werte > 3mg/24h sprechen für Phäochromozytom. MAO-Hemmer, Resperin, Alpha-Methyldopa, Tetrazykline, Beta-Blocker, etc. sollten vorher abgesetzt werden; auch Bananen, Zitrusfrüchte, Nüsse, Kaffee, Vanille, aber auch körperliche Belastung, Schock, Hypoglykämie, etc. können zu falsch-positiven Werten führen.
→ 2) Bei Verdacht auf ein malignes Phäochromozytom sollte zusätzlich noch Dopamin und Homovanillinsäure mitbestimmt werden.
→ IV: Bestätigungstest: (Als Clonidin-Hemmtest) Nach Gabe von 300µg Clonidin per os (= Alpha-2-Rezeptoragonist) sollte durch die zentrale Hemmung der Katecholaminausschüttung die Plasmakonzentration der Katecholaminmetaboliten sinken. Diese Konzentrationsabnahme findet man jedoch nicht bei einer autonomer Katecholaminsekretion wie beim Phäochromozytom.
→ V: Glukagontest: (= Provokationstest) Dieses Verfahren wird nur in Ausnahmefällen durchgeführt, wenn die vorangegangenen Untersuchungen uneindeutig waren, da mit dem Glukagontest schwere hypertensive Krisen ausgelöst werden können. Das Prinzip beruht darauf, dass Glukagon bei Patienten mit Phäochromozytom die Freisetzung der Katecholamine stimuliert. Nach Applikation von 1mg Glukagon erfolgt eine Blutentnahme nach 2, 5 und 10 Minuten mit Bestimmung der Katecholaminkonzentration. Ein Noradrenalinanstieg von > 300% ist immer pathologisch und weist auf ein Phäochromozytom hin. (Während der Untersuchung sind eine engmaschige Blutdruck-Kontrolle sowie die Bereitstellung von blutdrucksenkenden Medikamenten wie Alpha-Blocker im Notfall obligat).
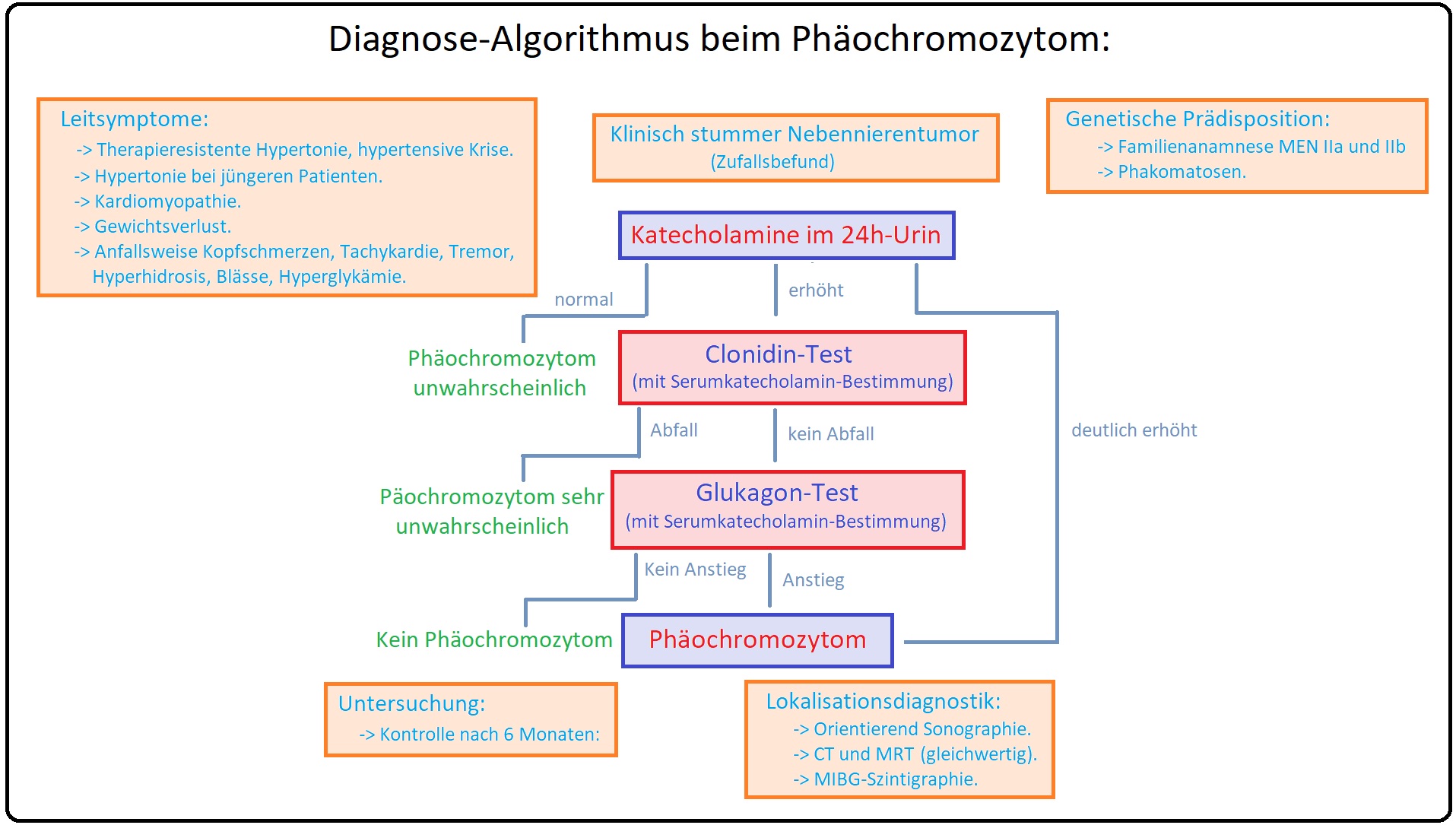
→ VI: Bildgebende Verfahren: Dient der Lokalisationsdiagnostik; dabei ist jedoch zu beachten, dass die Einteilung einer medikamentösen Therapie vor der Untersuchung zur Vermeidung einer hypertensiven Krise obligat ist.
→ 1) CT-/MRT-Abdomen: Indiz ist eine Vergrößerung der gesamten Nebenniere ohne Möglichkeit der Differenzierung zwischen Rinde und Mark.
→ 2) SPECT: (= Single-Photon-Emissions-CT) mit 123Jod-MIBG (Metajodbenzylguanidin) zur Lokalisationsdiagnostik bei kleinen extraadrenal gelegenen Tumoren, ggf. Somatostatin-Rezeptor-Szintigraphie bei Tumorverdacht.
→ VII: Histologie: In den meisten Fälle ist eine histologische Unterscheidung zwischen benigne und maligne kaum möglich. In diesem Zusammenhang existieren Malignitätskriterien wie lokale Invasion (insbesondere Gefäßeinbrüche) sowie der Nachweis von Fernmetastasen in z.B. Lymphknoten, Lunge, Leber und Skelett. Zudem lassen sich in den betroffenen Zellen Chromogranin A und neurospezifische Endolase (NSE) eruieren.
→ VIII: Genetische Diagnostik: Bei MEN Typ IIa.
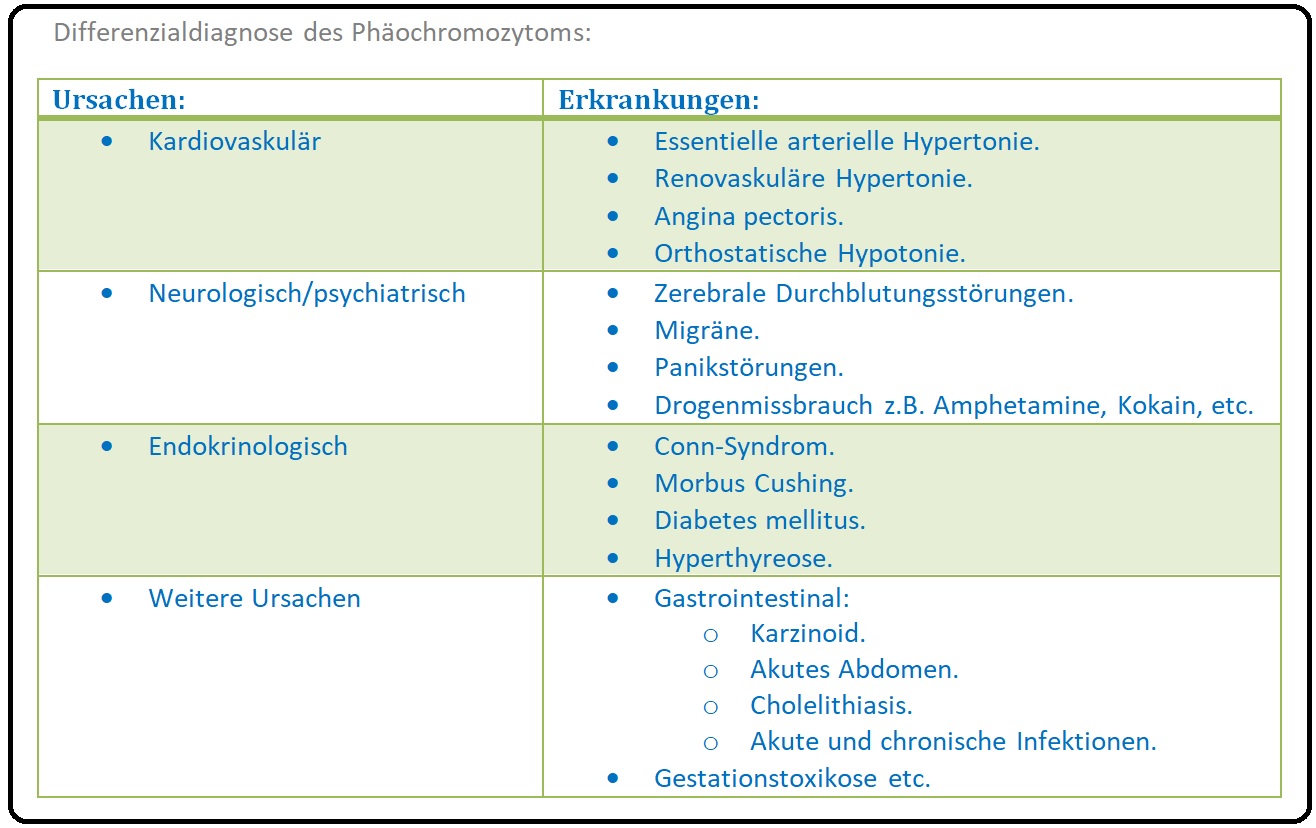
→ Differenzialdiagnose: Vom Phäochromozytom müssen insbesondere nachfolgende Erkrankungen abgegrenzt werden:
→ I: Hypertensive Krise anderer Genese,
→ II: Hyperglykämie bei Diabetes mellitus,
→ III: Karzinoid-Syndrom,
→ IV: Hyperthyreose (Morbus Basedow, Schilddrüsen-Autonomie),
→ V: Psychische Störungen wie Angst- und Paniksstörungen (insbesondere die Panikattacke) sowie Kokain- und Amphetaminabusus.
→ Therapie:
→ I: Konservative Therapie:
→ 1) Therapie der hypertensiven Krise (Siehe hypertensive Krise/ hypertensiver Notfall).
→ 2) Bei Inoperabilität Gabe eines Alpha-Blockers (Phenoxyzosin, Prazosin).
→ 3) Metastasiertes Phäochromozytom: Bei 123MIBG-(Metajodbenzylguanidin) positiven Metastasen ist eine Behandlung mit 123Jod-MIBG indiziert; bei Knochenmetastasen erfolgt eine palliative Bestrahlung; die Tumoren sprechen nur schlecht auf eine Chemotherapie an.
→ II: Operative Therapie:
→ 1) Präoperativ: ist die Vorbehandlung mit einem Alpha-Rezetor-Blocker (z.B. Phenoxybenzamin, Phenoxyzosin, Prazosin) und eine Volumenauffüllung obligat, um einer postoperativen Vasodilatation mit konsekutivem Blutdruckabfall und Kreislaufkollaps entgegenzuwirken (mit der Entfernung des Tumors entfällt der Induktor der chronischen Vasokonstriktion).
→ Klinisch-relevant: Man beginnt einschleichend mit einer Anfangsdosis von 10mg Phenoxybenzamin über 1-2 Wochen und steigert anschließend auf eine Dosis von 60-120 mg/d abhängig von der arteriellen Hypertonie bzw. der Höhe der Katecholaminkonzentration. Manifestiert sich zusätzlich ein Herzfrequenzanstieg ist die Gabe eines Beta-Blockers indiziert.
→ 2) Bei der unilateralen Form erfolgt eine einseitige laparoskopische Adrenalektomie (En-bloc-Resektion), bei der bilateraler Form (MEN) ist eine subtotale Adrenalektomie mittels No-Touch-Technik (ansonsten besteht die Gefahr der Katecholaminausschüttung) indiziert. Ein postoperativ induzierter Morbus Addison infolge einer bilateralen Adrenalektomie wird durch die lebenslange Glukokortikoid- und Mineralkortikoid-Substitution behandelt. Postoperativ muss das Risiko einer Hypoglykämie beachtet werden.
→ III: Nachsorge: Kontrolluntersuchungen mit Bestimmung der Katecholamine sind nach Tumorresektion obligat:
→ 1) Im ersten Jahr alle 3 Monate.
→ 2) Im zweiten Jahr alle 6 Monate und
→ 3) Im dritten Jahr jährlich.
→ Prognose: 50% der Patienten mit Phäochromozytom (benigne) sind postoperativ normotensiv. 15% der Patienten bilden jedoch ein Rezidiv mit Gefahr der malignen Entartung aus (die 5-Jahresüberlebenschance liegt beim malignen Phäochromozytom bei ca. 40%); Folglich sind regelmäßige Kontrolluntersuchungen indiziert:
→ I: Bestimmung der Plasmametanephrine und Chromogranin A (2 Monate postoperativ, anschließend jährlich).
→ II: Jährliche Abdomen-Sonographie.