→ Definition: Das medulläre Schilddrüsenkarzinom ist ein von den medullären kalzitoninproduzierenden C-Zellen der Schilddrüse ausgehender maligner Tumor mit vermehrter Produktion des Peptidhormons, Kalzitonin (Chromogranin A, Somatostatin, CEA). Als Rarität existiert zudem das atypische medulläre Schilddrüsenkarzinom ohne immunhistochemisch nachweisbares Kalzitonin und/oder ohne serologisch eruierbares erhöhtes Kalzitonin.
→ Epidemiologie:
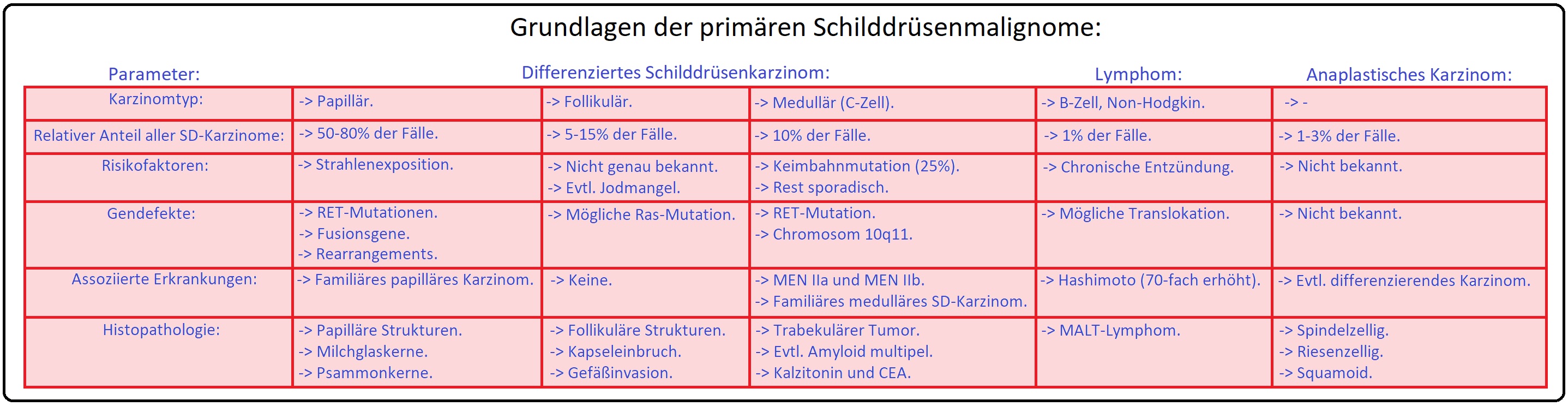
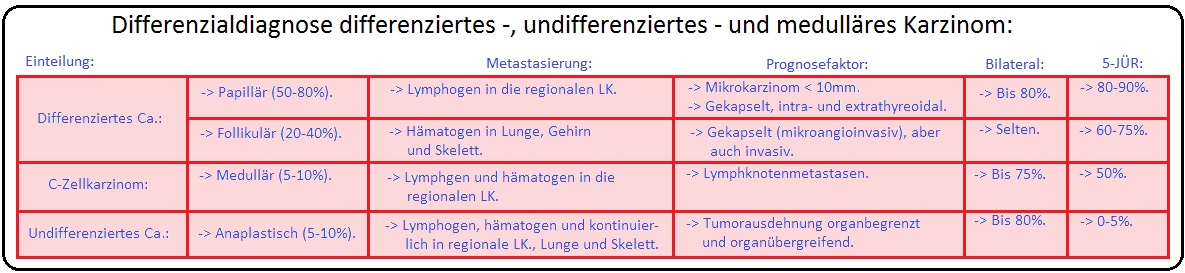
→ I: Das medulläre Schilddrüsenkarzinom macht 3-5% der malignen Schilddrüsentumoren aus; die Inzidenz liegt bei 0,1-0,2/1000000 Einwohnern pro Jahr und stellt somit eine sehr seltene Erkrankung dar.
→ II: Der Manifestationsgipfel liegt zwischen dem 40.-70. Lebensjahr (kann aber in jedem Lebensalter auftreten), wobei Männer nahezu genauso häufig betroffen sind wie Frauen. Beim familiär gehäuft auftretenden C-Zell-Karzinom liegt das Durchschnittsalter zwischen dem 15.-20. Lebensjahr.
→ Ätiologie: Man unterscheidet hierbei 2 Formen:
→ I: Sporadische Form: Mit 75% die häufigere Form; der Manifestationsgipfel liegt zwischen dem 50.-60. Lebensjahr. In 30-70% der Fälle zeigt sich hierbei eine somatische Mutation im Bereich des Kodons 918 (sie ist mit einer schlechten Prognose assoziiert).
→ II: Familiäre Form: (25%) Wird autosomal-dominant vererbt mit Mutationen des RET-Proto-Onkogens (kodiert ein membranständiges Protein mit einer Tyrosinkinase-Aktivität) auf Chromosom 10 (10q11.2) und Nachweis eines C-Zell-Karzinoms im Jugendalter (Die Penetranz des MTC liegt bei anhezu 100%). In den meisten Fällen tritt es im Zusammenhang mit dem MEN II (A oder B)-Syndrom auf, kann sich aber auch selten isoliert entwickeln.
→ Histopathologie: Histopathologische handelt es sich beim medullären Schilddrüsenkarzinom um einen nicht abgekapselten Tumor, der ein läppchenartiges bis trabekuläres Wachstum aufweist und sich aus spindeligen oder runden, polygonalen Zellen zusammensetzt. Bemerkenswert bei diesem Tumor ist, dass er die Fähigkeit besitzt, das Wachstumsmuster der follikulären -, papillären - bzw. anaplastischen Karzinome zu imitieren und in diesem Zusammenhang z.T. nur mit Hilfe der Immunhistochemie durch Nachweis der Kalzitoninsynthese differenziert werden kann. Die im Rahmen einer MEN II auftretenden medullären Schilddrüsenkarzinome treten typischerweise bilateral sowie multizentrisch auf und entwickeln sich aus einer C-Zell-Hyperplasie (= neoplastische Vorstufe). Der Manifestationsgipfel liegt insbesondere zwischen dem 6. Lebensmonat und 10. Lebensjahr (Abb.: Histologischer Aufbau des Schilddrüsengewebes).

→ Klinik:
→ I: Das medulläre Schilddrüsenkarzinom ist lange Zeit asymptomatisch und meist eine Zufallsdiagnose im Zuge einer SD-Untersuchung bei nodulärer Struma (siehe auch follikuläres/papilläres SD-Karzinom).
→ II: Symptome entstehen erst durch das infiltrative Wachstum des Malignoms:
→ 1) Lokale Zeichen: Höckriger, derber Knoten, regionale, nicht-schmerzhafte Lymphknotenschwellung, fixierte Haut.
→ 2) Durch Infiltration in die Nachbarorgane manifestiert sich u.a Stridor, Schluckbeschwerden, Heiserkeit (infolge einer Recurrensparese) und evtl. das Horner-Syndrom (mit Miosis, Ptosis und Endophthalmus).
→ III: Charakteristikum beim medullären Schilddrüsenkarzinom ist die Entwicklung einer therapieresistenten Diarrhoe durch Bildung vasoaktiver Hormone.
→ IV: Metastasierung: Das medulläre Schilddrüsenkarzinom metastasiert frühzeitig in die regionären Lymphknoten, erst sekundär kommt es zur einer hämatogenen Streuung. Fernmetastasen findet man insbesondere in:
→ 1) Skelett (40% der Fälle),
→ 2) Lunge (25% der Fälle) und
→ 3) Leber.
→ Diagnose:
→ I: Anamnese: MEN in der Familie; häufig liegen jedoch keine Beschwerden vor. Bei ausgedehnter Fernmetastasierung (u.a. in Lunge, Leber, Knochen, etc.) können sich als Folge eines Hormonexzesses bzw. einer paraneoplastischen Hormonproduktion eine chronische Diarrhö, Flush, ein ACTH-abhängiger Morbus Cushing sowie SIADH manifestieren.
→ II: Labor:
→ 1) Bestimmung der Kalzitonin-Konzentration. Typischerweise ist diese mehrfach bis tausend-fach erhöht, parallel hierzu besteht zusätzlich ein Anstieg von CEA (= Carzinoembryonales Antigen).

→ Klinisch-relevant:
→ A) Die Kalzitonin-Serumkonzentration korreliert mit der Tumorlast (Durchmesser des Primärtumors, Lymknotenfiliae, Fernmetastasen).
→ B) Die Kalzitonin- und CEA-Verdopplungszeiten wiederum korrelieren mit der Tumorprogression und Gesamtüberlebenszeit.
→ 2) Pentagastrin-Test: Zur Frühdiagnose oder postoperativ zur Rezidivkontrolle. Die Gabe von 0.5µg/kgKG Pentagastrin i.v. verursacht nach 3-5 min eine vermehrte Sekretion von Kalzitonin. Beim MTC steigt die Kalzitonin-Konzentration > 5-fache an.
→ III: Bildgebung:
→ 1) Sonographie: Insbesondere echoarme intrathyreoidale Raumforderungen sowie mögliche zervikale malignitätsverdächtige Lymphknoten.
→ 2) Zur Metastasensuche dienen CT und MRT, aber auch die FDG-PET sowie die Somatostatinrezeptor-Szintigraphie finden zunehmend Bedeutung.
→ IV: C-Zell-Karzinom: Genanalyse auf Punktmutation des Protoonkogens und familiäres Screening (MEN). Bei Familienangehörigen (insbesondere Kindern) von Patienten mit medullärem Schilddrüsenkarzinom sollte eine molekulargenetische Untersuchung mit Hinblick auf Punktmutationen des RET-Proto-Onkogens auf Chromosom 10 erfolgen.
→ Differenzialdiagnose: Calcitonin-Erhöhungen manifestieren sich unter anderem auch bei einer Pankreatitis, Niereninsuffizienz oder evtl. im Zuge eines paraneoplastischen Syndroms.
→ Therapie:
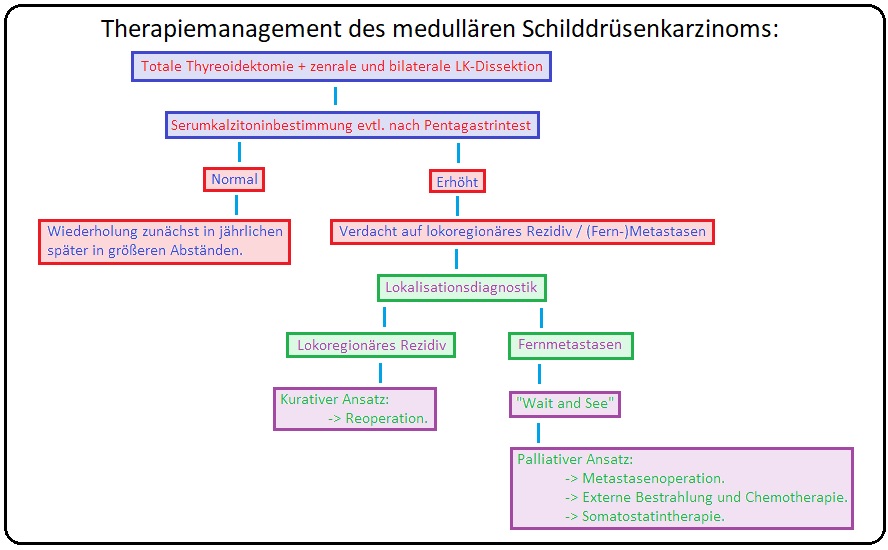
→ I: Operativ: (= Modifizierte Neck-Dissection) Mittel der Wahl ist die (radikale) totale Thyreoidektomie mit gleichzeitiger operativer Entfernung der zentralen und beider lateralen Lymphknotenkompartimente (evtl. auch der mediastinalen LK). Bleibt postoperativ die Kalzitoninkonzentration weiterhin erhöht, erfolgt eine nochmalige Tumor-/Metastasensuche mittels Röntgen-Thorax, Sonographie des Abdomens, CT, Somatostatinrezeptor-Szintigraphie, etc.
→ II: Bei fortgeschrittenem Tumorstadium: Führt eine Therapie mit Tyrosinkinase-Inhibitoren in 50% zur partiellen Tumorremission.
→ III: Systemische Chemotherapie: Die Kombinationstherapie bestehend aus Cyclophosphamid, Vincristin und Doxorubicin stellt eine weitere Behandlungsoption dar.
→ IV: Bei MEN-II-Syndrom: Prophylaktische Thyreoidektomie und regelmäßige Kontrolluntersuchungen auf Phäochromozytom und primären Hyperparathyreoidismus.
→ V: Nachsorge:
→ 1) Regelmäßige halbjährige Kontrolluntersuchungen mit Bestimmung des Kalzitonin- und CEA-Wertes.
→ 2) Routinemäßige bildgebende Verfahren zur Tumorsuche bzw. Rezidivprophylaxe.
→ Klinisch-relevant:
→ A) Eine Radiojodtherapie ist beim medullären SD-Karzinom, aufgrund der fehlenden Jodspeicherung, unwirksam.
→ B) Eine perkutane Strahlentherapie ist wegen der Strahlenresistenz des Malignoms nicht indiziert.
→ Prognose: Die 5-Jahresüberlebenschance liegt beim medullären Schilddrüsenkarzinom insgesamt bei 75% (ohne Lymphknotenmetastasen bei 85%, mit Lk-Metastasen bei 42%). Wichtige prognostische Faktoren sind insbesondere Tumorstadium zum Zeitpunkt der Diagnose, Kalzitonin- und CEA-Verdopplungszeit (< 6 Monate prognostisch ungünstig; > 2 Jahre prognostisch günstig), Alter sowie der Form (sporadisch oder familiär) des medullären Schilddrüsenkarzinoms.