- Details
- Geschrieben von: CF
- Kategorie: Schilddrüse und Nebenschilddrüse
- Zugriffe: 4747
→ Definition: Unter dem Begriff Hyperthyreose werden die durch den vermehrten Schilddrüsengehalt in der Peripherie mit konsekutiver Stimulation der Stoffwechselvorgänge und sympathoadrenerger Überempfindlichkeit ausgelösten Krankheitsbeschwerden zusammengefasst.
→ Epidemiologie:
→ I: Die Hyperthyreose tritt im Erwachsenenalter (Allgemeinbevölkerung) mit einer Prävalenz von 1-2% auf.
→ II: In Deutschland wird sie zu gleichen Anteilen der Immunhyperthyreose (Morbus Basedow) und der Schilddrüsenautonomie zugeteilt, während in Ländern mit ausreichender Jodversorgung die Autonomie in den Hintergrund tritt.
→ III: Frauen sind 5-8x häufiger von einer Hyperthyreose betroffen als Männer, wobei der Manifestationsgipfel zwischen dem 25.-60. Lebensjahr liegt.
→ Ätiologie: Die häufigsten Ursachen sind insbesondere die Schilddrüsenautonomie aufgrund eines Jodmangels sowie die Immunhyperthyreose. Letztere gilt heute als genetisch determinierte Störung des Immunsystems, die sich durch:
→ I: Hyperaktivität des lymphatischen Systems.
→ II: Lymphozytäre Infiltration des Schilddrüsenparenchyms,
→ III: Häufiger Nachweis von Schilddrüsenautoantikörpern
→ IV: Assoziation zu HLA-Antigenen und nicht zuletzt
→ V: Aufgrund des gehäuften familiären Vorkommens bzw. der Assoziation zu anderen Autoimmunerkrankungen.

→ Pathogenese: Nachfolgende Effekte sind allen Formen der Hyperthyreose gemeinsam und basieren auf erhöhten Schilddrüsenhormonen im Organismus mit konsekutiver Steigerung der schilddrüsenhormonabhängigen biologischen Prozesse:
→ I: Steigerung der Oxidation, Thermogenese, des Cholesterin- und Glykogenabbaus sowie des Umsatzes der freien Fettsäuren.
→ II: Steigerung des Proteinumbaus (beinhaltet sowohl die Synthese als auch den Abbau, wobei letzteres bei der Hyperthyreose überwiegt).
→ III: Katecholaminwirkung: Bei bestehender Hyperthyreose manifestiert sich eine erhöhte Sensibilität gegenüber den Katecholaminen mit:
→ 1) Gesteigerter Herzfrequenz,
→ 2) Gesteigerter Muskelkontraktion sowie
→ 3) Erhöhter Nervenerregbarkeit.
→ 4) Zusätzlich zeigt sich ein beschleunigter Abbau der Nebennierenrindenhormone (Mineralkortikoide, Glukokortikoide, Androgene).
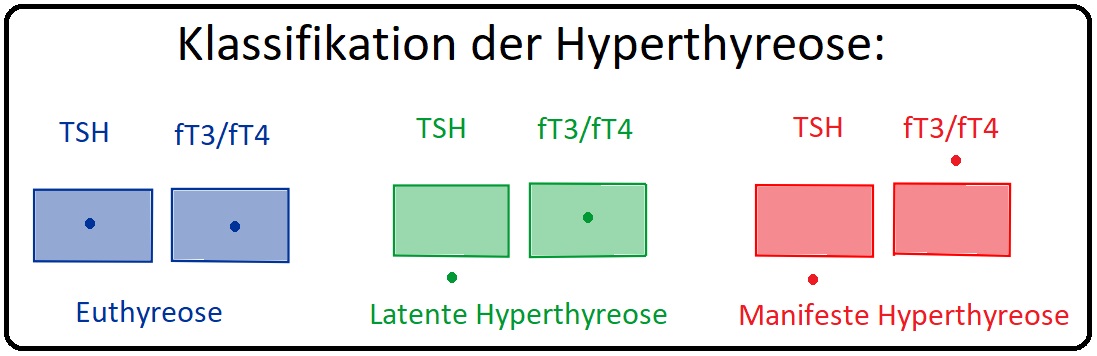
→ Klassifikation: Die Hyperthyreose wird nach ihrem Schweregrad eingeteilt in:
→ I: Latente Hyperthyreose: Bei dieser Form ist TSH supprimiert, jedoch liegen die peripheren Schilddrüsenhormone im Normbereich.
→ II: Manifeste Hyperthyreose: Hier ist neben dem TSH auch die peripheren Schilddrüsenhormone erhöht und es besteht in der Regel eine klinische Symptomatik.
→ III: Die thyreotoxische Krise stellt eine lebensbedrohliche endokrine Notfallsituation dar.
→ Klinik: Die klinische Symptomatik der Hyperthyreose resultiert unabhängig von der Genese (z.B. Schilddrüsenautonomie, Morbus Basedow, Hashimoto-Thyreoiditis, Thyreoiditis de Quervain, etc.) aus der Wirkung der Schilddrüsenhormone in der Peripherie.
→ I: Klinische Leitsymptome sind u.a.:
→ 1) Allgemeinsymptome mit innerer Unruhe, Nervosität, Schlafstörungen, vermehrtes Schwitzen, Wärmeintoleranz, aber auch depressive Episoden (insbesondere im höheren Lebensalter), etc.
→ 2) Kardiale Symptome: Mit Ruhetachykardie, Palpitation, Tachyarrhythmie bis hin zum Vorhofflimmern.
→ 3) GIT-Beschwerden: Mit vermehrtem Appetit, erhöhter Stuhlfrequenz bis hin zur Diarrhö.
→ II: Insbesondere im höherem Lebensalter (nach dem 60. Lebensjahr) sind die Beschwerden zumeist nur mono- oder oligosymptomatisch.
→ III: Bei Dekompensation einer schweren Hyperthyreose kann sich eine thyreotoxische Krise entwickeln, die auch heute noch häufig letal endet.

→ Diagnose: Die Diagnose der manifesten Hyperthyreose ergibt sich in Abhängigkeit von der Form der Hyperthyreose aus der Anamnese, klinische Untersuchung, Laborparametern und bildgebender Diagnostik.
→ I: Anamnese: Familiäre Häufung von Schilddrüsenerkrankungen, Medikamentenanamnese (z.B. Amiodaron, etc.), Eruierung der Kardinalssymptome (z.B. Nervosität, Schwitzen, Ruhetachykardie, Gewichtsabnahme, etc.).
→ II: Klinische Untersuchung: Inspektion und Palpation der Schilddrüse mit möglicher Erfassung von:
→ 1) Grad der Struma (I-III) inklusive Bestimmung des Halsumfangs.
→ 2) Knoten (Größe, Schmerzhaftigkeit, Konsistenz, Schluckverschieblichkeit).
→ 3) Stridor, Schwirren, Halsvenenstauung und Erfassung des cervikalen Lymphknotenstatus.
→ 4) Allgemeine Untersuchung mit Puls, Blutdruck, Ödemen, etc.
→ III: Labor:
→ 1) Das bedeutendste Laborparameter in der Hyperthyreose-Diagnostik ist das TSH basal sowie der freien T3/T4 Schilddrüsenhormone.
→ 2) Schilddrüsenautoantikörper: Sie dienen der Differenzialdiagnose einer Hyperthyreose mit Nachweis autoimmunologischer Formen. Die Bestimmung der Autoantikörper sollte im Stadium der floriden Hyperthyreose erfolgen und umfasst die TSH-Rezeptor-Antikörper (TRAK), Thyreogobulin-Antikörper (TAK) und mirkosomale AK (MAK).
→ IV: EKG: Die EKG-Veränderungen beruhen fast ausschließlich auf der Wirkung der Schilddrüsenhormone auf den Herzmuskel selbst:

→ V: Bildgebende Diagnostik:
→ 1) Sonographisch werden Aussagen über Größe, Lage und nicht zuletzt Struktur des Organs getroffen. Häufig zeigt sich bei der Hyperthyreose eine ausgeprägte Echoarmut des Schilddrüsenparenchyms.
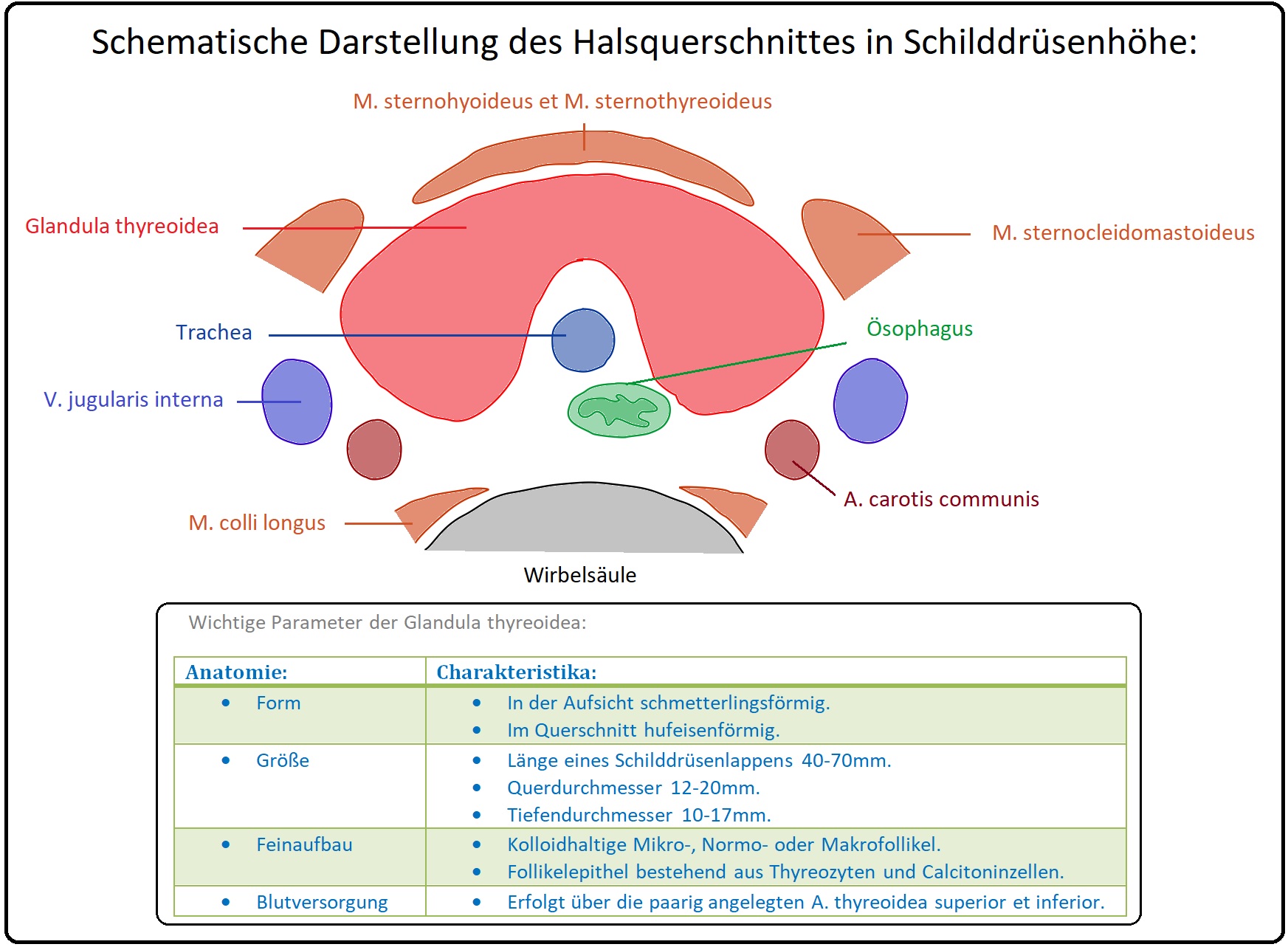
→ 2) Die Schilddrüsen-Szintigraphie dient als ergänzendes Diagnostikum zur Darstellung morphologischer Besonderheiten.
→ Differenzialdiagnose: Die Differenzialdiagnosen des klinischen Krankheitsbildes sind breit gefächert und umfassen:
→ I: Vegetative Dystonie, aber auch Wechseljahre,
→ II: Kardiale Erkrankungen: Wie u.a.:
→ 1) Sick-Sinus-Syndrom,
→ 2) Herzrhythmusstörungen bis hin zum Vorhofflimmern,
→ 3) Koronare Herzkrankheit,
→ 4) Kardiomyopathie, etc.
→ III: Diabetes mellitus,
→ IV: B-Symptomatik bei malignen Neoplasien.
→ V: Myasthenia gravis (Ermüdbarkeit, Adynamie, periodische Paralysen).
→ VI: Psychischtrische Erkrankungen: Wie z.B.
→ 1) Psychosen anderer Genese,
→ 2) Involutionsdepression, aber auch
→ 3) Chronische Alkoholabhängigkeit und nicht zuletzt das
→ VII: Phäochromozytom.
→ Therapie: Allgemein hängt die Therapiewahl entscheidend von der Form der Hyperthyreose ab und es stehen prinzipiell 3 Behandlungssäulen zur Verfügung.
→ I: Medikamentöse Therapie: (= thyreostatisch)
→ 1) Hierbei stehen verschiedene Medikamente zur Verfügung, wobei ist Carbimazol bzw. Thiamazol sich als Mittel der ersten Wahl herauskristallisiert haben. Diese Thyreostatika hemmen die Bildung der Schilddrüsenhormone durch Blockade der Umwandlung des Jodthyrosins zu Jodthyronin.
→ 2) Eine Alternative bei z.B. Unverträglichkeit der Thyresotatika vom Mercaptoimidazol-Typ stellt Propylthiouracil mit gleichem biochemischen Angriffspunkt dar.
→ 3) Perchlorat hemmt die Aufnahme von Jodid in die Schilddrüse und wird insbesondere zum prophylaktischen Schutz der Schilddrüse z.B. bei SD-Autonomie vor Jodexposition eingesetzt.
→ Klinisch-relevant:
→ A) Das Blutbild während der Thyreostatikatherapie sollte zu Behandlungsbeginn eine engmaschige Laborkontrolle zum frühzeitigen Nachweis einer möglichen Leukozytopenie bzw. Agranulozytose, im weiteren Therapieverlauf in ¼-jährigen Abständen erfolgen.
→ B) Der Schilddrüsenhormonspiegel sollte unter der medikamentösen Behandlung nicht zu tief absinken, um eine vermehrte TSH-Sekretion mit Gefahr der Struma-Bildung zu vermeiden.
→ II: Operative Therapie: Sie dient der Resektion des überaktiven Schilddrüsenparenchyms.
→ III: Radiojodtherapie: Diese Behandlungsoption stellt eine Alternative zur chirurgischen Intervention dar (sie ist während einer Schwangerschaft kontraindiziert und im Anschluss an eine 6 Monate dauernde Karenzphase möglich).
→ Prognose: Die klinische Symptomatik der Hyperthyreose sistiert unter der Therapie mit Erreichen einer euthyreoten Stoffwechsellage rasch.
- Details
- Kategorie: Schilddrüse und Nebenschilddrüse
- Zugriffe: 11345
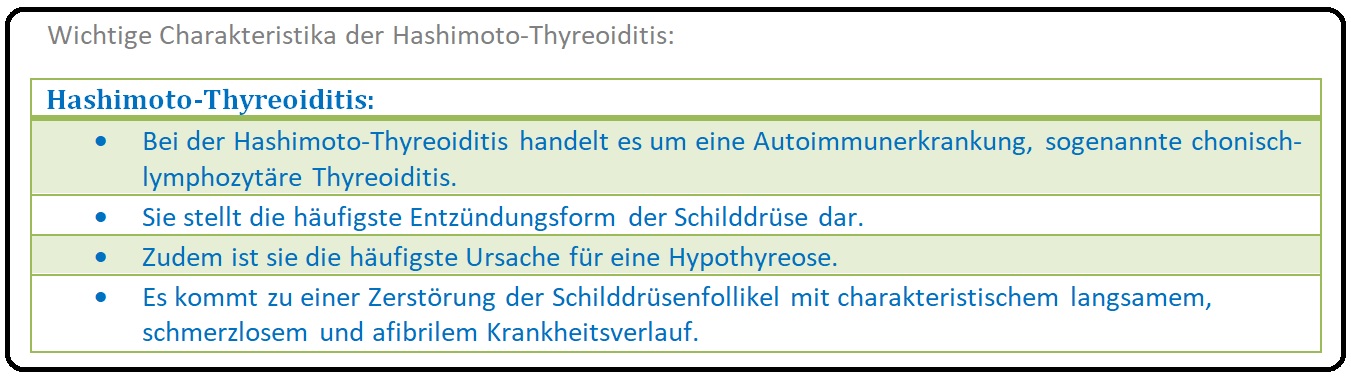
→ Definition: Bei der Hashimoto-Thyreoiditis handelt es sich um eine chronisch-lymphozytäre Schilddrüsenentzündung autoimmuner Genese und geht definitionsgemäß mit einer vergrößerten Schilddrüse (= Struma lymphomatosa) einher.
→ Epidemiologie:
→ I: Sie ist die häufigste Form der chronischen Thyreoiditis und die häufigste Ursache einer Hypothyreose.
→ II: Frauen sind 9 x häufiger als Männer betroffen, wobei der Manifestationsgipfel zwischen dem 30.-50. Lebensjahr liegt.
→ III: Sie ist häufig mit anderen Autoimmunerkrankungen, wie z.B. der Morbus Addison, Diabetes mellitus Typ I, Hypoparathyreoidismus und nicht zuletzt mit dem polyendokrinen Autoimmun-Syndrom vergesellschaftet.
→ Ätiologie:
→ I: Familiäre Disposition mit bis zu 50% der Fälle bei Verwandten.
→ II: HLA-DR3/DR4 assoziiert mit Vitiligo und Alopezie.
→ III: Gehäuftes Auftreten bei Hepatitis-C.
→ Pathogenese: Der Befall des Schilddrüsengewebes durch T-Lymphozyten und Plasmazellen (TPO-Ak, TgAk) führt im Laufe der Zeit zu einer Zerstörung der Thyreozyten mit konsekutiver Ausbildung einer primären Hypothyreose. Histologisch zeigt sich eine lymphozytäre Entzündung, die im weiteren Krankheitsverlauf zur Fibrose und Atrophie des Schilddrüsengewebes führt.
→ Klinik:
→ I: Die chronische Hashimoto-Thyreoiditis verläuft lange Zeit asymptomatisch in Form einer schmerzlosen Entzündung.
→ II: Initial kann sich eine Hyperthyreose (= Hashitoxikose) mit ihren typischen Symptomen, aufgrund der autoimmunologischen Zerstörung der Thyreozyten mit nachfolgender vermehrter Hormonfreisetzung, ausbilden.
→ III: Erst im Spätstadium entwickeln sich, aufgrund des zunehmenden Thyreozytenverlustes infolge entzündlich- lymphozytärer Destruktionsprozesse, die Charakteristika einer Hypothyreose mit allgemeiner Schwäche, zunehmender Apathie, Kälteintoleranz, Gewichtszunahme, Obstipation, teigiger Haut, Bradykardie, weiteren psychischen Veränderungen bis hin zur Depression etc.
→ Diagnose:
→ I: Labor:
→ 1) TSH-basal erhöht, T3/T4 erniedrigt. Sind die Schilddrüsenhormone im Normbereich spricht man von einer latenten Hypothyreose.
→ 2) Nachweis von Auto-AK gegen die thyreoidale Peroxidase (= TPO-AK = mikrosomale AK = MAK) in 95%.
→ 3) Nachweis von AK gegen Thyreoglobulin (= TgAK) in 70% der Fälle.
→ IV: Sonographie: Die Schilddrüse stellt sich inhomogen, echoarm und meist verkleinert dar.
→ V: SD-Szintigraphie: Verminderte Radionukleotid-Anreicherung in der SD.

→ Differenzialdiagnose:
→ I: Morbus Basedow: Nachweis von TSH-Rezeptor-AK (= TRAK > 95%) und Thyreoglobulin AK in 70%.
→ II: Hyperthyreose bei Schilddrüsenautonomie.
→ III: Silent-Thyreoditis: Milde Form evtl. auch nur temporär.
→ IV: Postpartale lymphgene Thyreoditis: In 4% manifestiert sich im Rahmen der Postpartalphase eine passagere oder latente SD-Funktionsstörung mit zumeist positiven Nachweis von Auto-AK gegen TPO.
→ V: Iatrogen verursachte Hypothyreose:
→ 1) Aufgrund einer Interleukin-2 oder Interferontherapie,
→ 2) Thyreostatika wie z.B. Thionamide und Perchlorat,
→ 3) Lithium-Therapie,
→ 4) Amiodaron-Therapie.
→ VI: Riedel-Struma: Als chronisch-fibrosierende Struma mit schwartenartigem, hart infiltierendem und ummauerndem Wachstum; sehr selten.
→ Therapie:
→ I: Indikationen: Folgende Situationen stellen eine Indikation für eine Substitutionstherapie dar:
→ 1) Manifeste Hypothyreose und
→ 2) Latente Hypothyreose mit klinischen Symptomen, wobei insbesondere auch auf den TSH-Wert > 10mU/l geachtet wird.
→ II: Medikamentöse Therapie:
→ 1) Lebenslange L-Thyroxin-Substitution bei vorhandener Hypothyreose. Die richtige Dosierung (1,5µg/kgKG/d) ist bei TSH Normalisierung (0,5-2mU/l) und Wohlbefinden des Patienten erreicht.
→ 2) Da L-Thyroxin eine HWZ von ca. 1 Woche hat, sollte die TSH-Kontrolle erst 4-6 Wochen nach Dosisanpassung erfolgen.
→ Klinisch-relevant: Bei älteren Menschen und Patienten mit KHK sollte mit einer niedrigen Dosierung begonnen werden und alle 2-4 Wochen gesteigert werden.
→ 3) Steroide zur Behandlung der Autoimmunthyreoiditis sind wirkungslos.
→ 4) Bei euthyreoter Autoimmunthyreoiditis kann die Applikation von Selen zur Senkung der Auto-AK versucht werden (Evidenz jedoch nicht bewiesen).
- Details
- Kategorie: Schilddrüse und Nebenschilddrüse
- Zugriffe: 13682
→ Definition: Das medulläre Schilddrüsenkarzinom ist ein von den medullären kalzitoninproduzierenden C-Zellen der Schilddrüse ausgehender maligner Tumor mit vermehrter Produktion des Peptidhormons, Kalzitonin (Chromogranin A, Somatostatin, CEA). Als Rarität existiert zudem das atypische medulläre Schilddrüsenkarzinom ohne immunhistochemisch nachweisbares Kalzitonin und/oder ohne serologisch eruierbares erhöhtes Kalzitonin.
→ Epidemiologie:
→ I: Das medulläre Schilddrüsenkarzinom macht 3-5% der malignen Schilddrüsentumoren aus; die Inzidenz liegt bei 0,1-0,2/1000000 Einwohnern pro Jahr und stellt somit eine sehr seltene Erkrankung dar.
→ II: Der Manifestationsgipfel liegt zwischen dem 40.-70. Lebensjahr (kann aber in jedem Lebensalter auftreten), wobei Männer nahezu genauso häufig betroffen sind wie Frauen. Beim familiär gehäuft auftretenden C-Zell-Karzinom liegt das Durchschnittsalter zwischen dem 15.-20. Lebensjahr.
→ Ätiologie: Man unterscheidet hierbei 2 Formen:
→ I: Sporadische Form: Mit 75% die häufigere Form; der Manifestationsgipfel liegt zwischen dem 50.-60. Lebensjahr. In 30-70% der Fälle zeigt sich hierbei eine somatische Mutation im Bereich des Kodons 918 (sie ist mit einer schlechten Prognose assoziiert).
→ II: Familiäre Form: (25%) Wird autosomal-dominant vererbt mit Mutationen des RET-Proto-Onkogens (kodiert ein membranständiges Protein mit einer Tyrosinkinase-Aktivität) auf Chromosom 10 (10q11.2) und Nachweis eines C-Zell-Karzinoms im Jugendalter (Die Penetranz des MTC liegt bei anhezu 100%). In den meisten Fällen tritt es im Zusammenhang mit dem MEN II (A oder B)-Syndrom auf, kann sich aber auch selten isoliert entwickeln.
→ Histopathologie: Histopathologische handelt es sich beim medullären Schilddrüsenkarzinom um einen nicht abgekapselten Tumor, der ein läppchenartiges bis trabekuläres Wachstum aufweist und sich aus spindeligen oder runden, polygonalen Zellen zusammensetzt. Bemerkenswert bei diesem Tumor ist, dass er die Fähigkeit besitzt, das Wachstumsmuster der follikulären -, papillären - bzw. anaplastischen Karzinome zu imitieren und in diesem Zusammenhang z.T. nur mit Hilfe der Immunhistochemie durch Nachweis der Kalzitoninsynthese differenziert werden kann. Die im Rahmen einer MEN II auftretenden medullären Schilddrüsenkarzinome treten typischerweise bilateral sowie multizentrisch auf und entwickeln sich aus einer C-Zell-Hyperplasie (= neoplastische Vorstufe). Der Manifestationsgipfel liegt insbesondere zwischen dem 6. Lebensmonat und 10. Lebensjahr (Abb.: Histologischer Aufbau des Schilddrüsengewebes).

→ Klinik:
→ I: Das medulläre Schilddrüsenkarzinom ist lange Zeit asymptomatisch und meist eine Zufallsdiagnose im Zuge einer SD-Untersuchung bei nodulärer Struma (siehe auch follikuläres/papilläres SD-Karzinom).
→ II: Symptome entstehen erst durch das infiltrative Wachstum des Malignoms:
→ 1) Lokale Zeichen: Höckriger, derber Knoten, regionale, nicht-schmerzhafte Lymphknotenschwellung, fixierte Haut.
→ 2) Durch Infiltration in die Nachbarorgane manifestiert sich u.a Stridor, Schluckbeschwerden, Heiserkeit (infolge einer Recurrensparese) und evtl. das Horner-Syndrom (mit Miosis, Ptosis und Endophthalmus).
→ III: Charakteristikum beim medullären Schilddrüsenkarzinom ist die Entwicklung einer therapieresistenten Diarrhoe durch Bildung vasoaktiver Hormone.
→ IV: Metastasierung: Das medulläre Schilddrüsenkarzinom metastasiert frühzeitig in die regionären Lymphknoten, erst sekundär kommt es zur einer hämatogenen Streuung. Fernmetastasen findet man insbesondere in:
→ 1) Skelett (40% der Fälle),
→ 2) Lunge (25% der Fälle) und
→ 3) Leber.
→ Diagnose:
→ I: Anamnese: MEN in der Familie; häufig liegen jedoch keine Beschwerden vor. Bei ausgedehnter Fernmetastasierung (u.a. in Lunge, Leber, Knochen, etc.) können sich als Folge eines Hormonexzesses bzw. einer paraneoplastischen Hormonproduktion eine chronische Diarrhö, Flush, ein ACTH-abhängiger Morbus Cushing sowie SIADH manifestieren.
→ II: Labor:
→ 1) Bestimmung der Kalzitonin-Konzentration. Typischerweise ist diese mehrfach bis tausend-fach erhöht, parallel hierzu besteht zusätzlich ein Anstieg von CEA (= Carzinoembryonales Antigen).

→ Klinisch-relevant:
→ A) Die Kalzitonin-Serumkonzentration korreliert mit der Tumorlast (Durchmesser des Primärtumors, Lymknotenfiliae, Fernmetastasen).
→ B) Die Kalzitonin- und CEA-Verdopplungszeiten wiederum korrelieren mit der Tumorprogression und Gesamtüberlebenszeit.
→ 2) Pentagastrin-Test: Zur Frühdiagnose oder postoperativ zur Rezidivkontrolle. Die Gabe von 0.5µg/kgKG Pentagastrin i.v. verursacht nach 3-5 min eine vermehrte Sekretion von Kalzitonin. Beim MTC steigt die Kalzitonin-Konzentration > 5-fache an.
→ III: Bildgebung:
→ 1) Sonographie: Insbesondere echoarme intrathyreoidale Raumforderungen sowie mögliche zervikale malignitätsverdächtige Lymphknoten.
→ 2) Zur Metastasensuche dienen CT und MRT, aber auch die FDG-PET sowie die Somatostatinrezeptor-Szintigraphie finden zunehmend Bedeutung.
→ IV: C-Zell-Karzinom: Genanalyse auf Punktmutation des Protoonkogens und familiäres Screening (MEN). Bei Familienangehörigen (insbesondere Kindern) von Patienten mit medullärem Schilddrüsenkarzinom sollte eine molekulargenetische Untersuchung mit Hinblick auf Punktmutationen des RET-Proto-Onkogens auf Chromosom 10 erfolgen.
→ Differenzialdiagnose: Calcitonin-Erhöhungen manifestieren sich unter anderem auch bei einer Pankreatitis, Niereninsuffizienz oder evtl. im Zuge eines paraneoplastischen Syndroms.
→ Therapie:
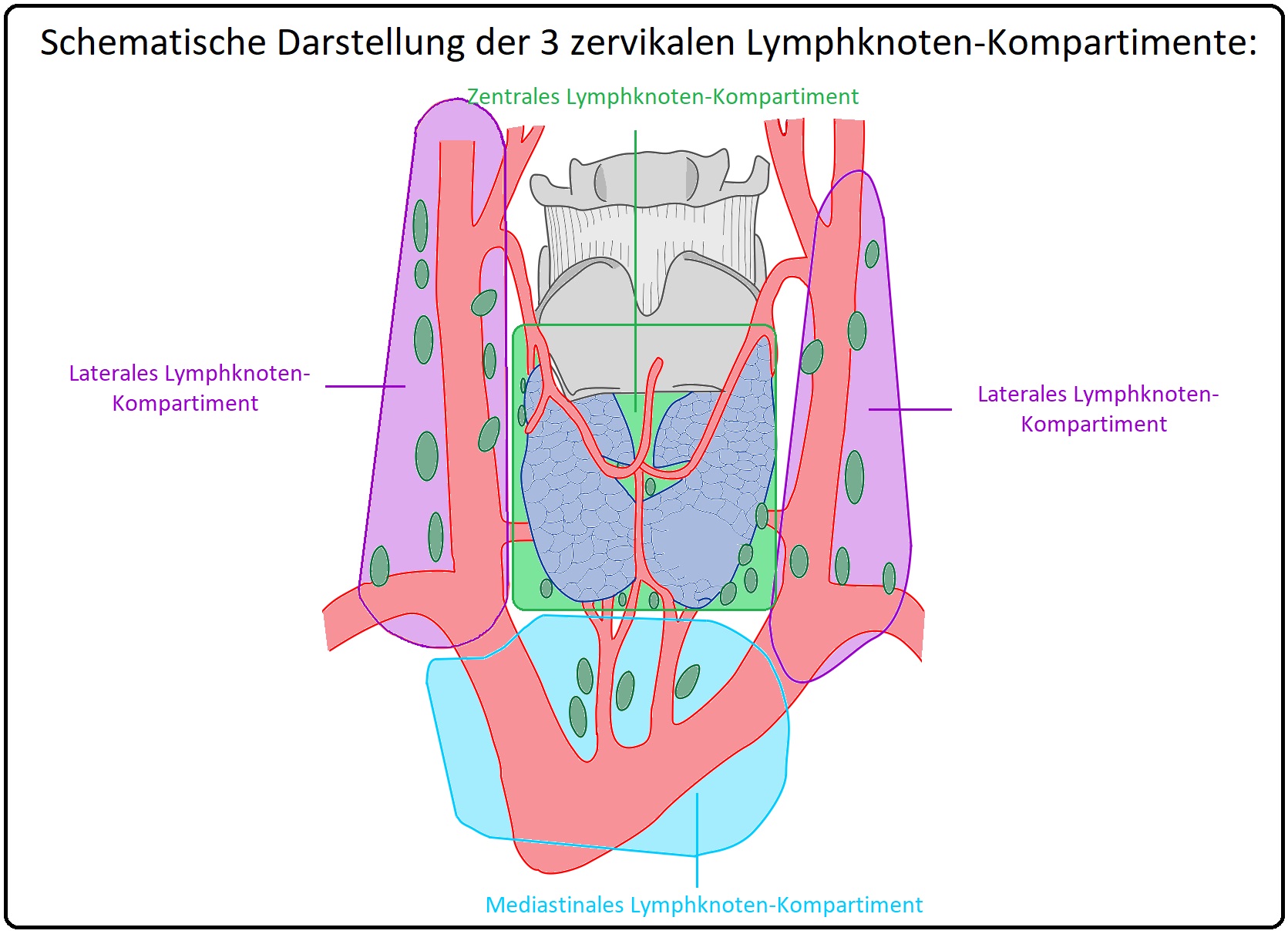
→ I: Operativ: (= Modifizierte Neck-Dissection) Mittel der Wahl ist die (radikale) totale Thyreoidektomie mit gleichzeitiger operativer Entfernung der zentralen und beider lateralen Lymphknotenkompartimente (evtl. auch der mediastinalen LK). Bleibt postoperativ die Kalzitoninkonzentration weiterhin erhöht, erfolgt eine nochmalige Tumor-/Metastasensuche mittels Röntgen-Thorax, Sonographie des Abdomens, CT, Somatostatinrezeptor-Szintigraphie, etc.
→ II: Bei fortgeschrittenem Tumorstadium: Führt eine Therapie mit Tyrosinkinase-Inhibitoren in 50% zur partiellen Tumorremission.
→ III: Systemische Chemotherapie: Die Kombinationstherapie bestehend aus Cyclophosphamid, Vincristin und Doxorubicin stellt eine weitere Behandlungsoption dar.
→ IV: Bei MEN-II-Syndrom: Prophylaktische Thyreoidektomie und regelmäßige Kontrolluntersuchungen auf Phäochromozytom und primären Hyperparathyreoidismus.
→ V: Nachsorge:
→ 1) Regelmäßige halbjährige Kontrolluntersuchungen mit Bestimmung des Kalzitonin- und CEA-Wertes.
→ 2) Routinemäßige bildgebende Verfahren zur Tumorsuche bzw. Rezidivprophylaxe.
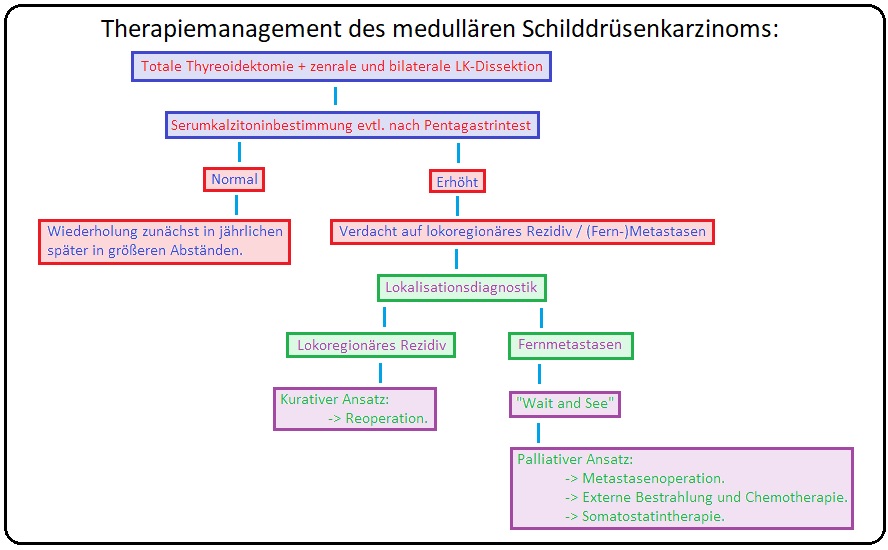
→ Klinisch-relevant:
→ A) Eine Radiojodtherapie ist beim medullären SD-Karzinom, aufgrund der fehlenden Jodspeicherung, unwirksam.
→ B) Eine perkutane Strahlentherapie ist wegen der Strahlenresistenz des Malignoms nicht indiziert.
→ Prognose: Die 5-Jahresüberlebenschance liegt beim medullären Schilddrüsenkarzinom insgesamt bei 75% (ohne Lymphknotenmetastasen bei 85%, mit Lk-Metastasen bei 42%). Wichtige prognostische Faktoren sind insbesondere Tumorstadium zum Zeitpunkt der Diagnose, Kalzitonin- und CEA-Verdopplungszeit (< 6 Monate prognostisch ungünstig; > 2 Jahre prognostisch günstig), Alter sowie der Form (sporadisch oder familiär) des medullären Schilddrüsenkarzinoms.
- Details
- Kategorie: Schilddrüse und Nebenschilddrüse
- Zugriffe: 12325
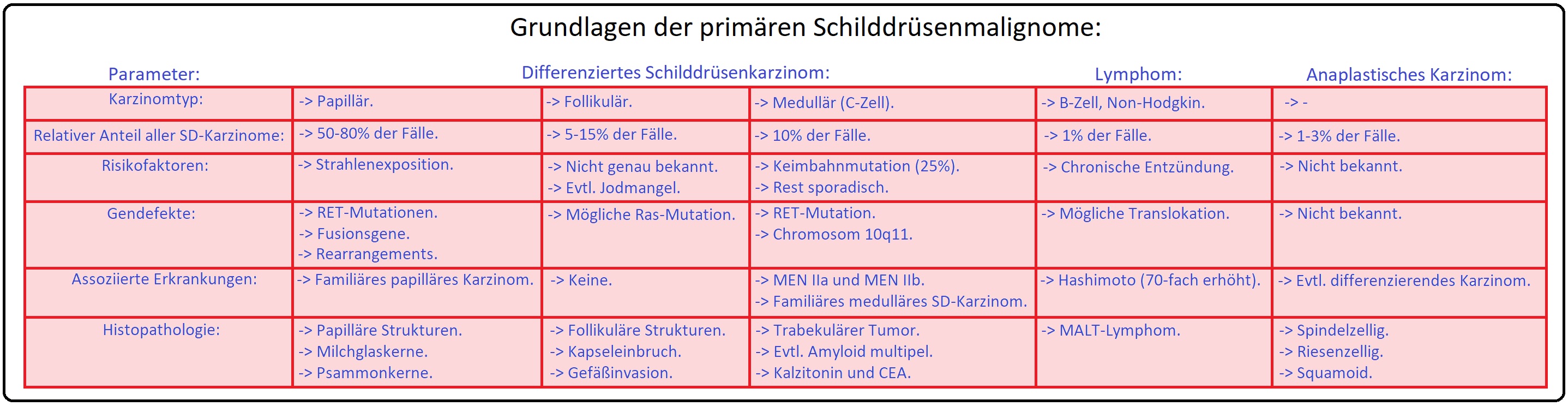
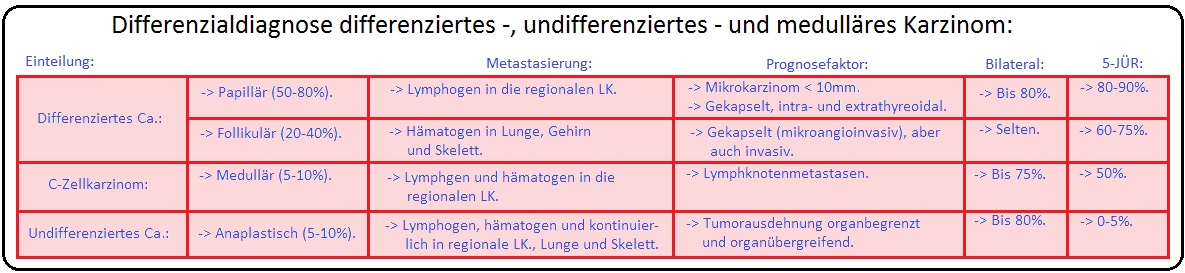
→ Definition: Das differenzierte Schilddrüsenkarzinom ist definiert als eine maligne, epitheliale Neoplasie, welche von den Thyreozyten der Schilddrüse ausgeht. Hierbei wird zwischen 2 Subtypen, dem papillären und follikulären Karzinom, unterschieden.
→ Epidemiologie:
→ I: Das Schilddrüsenkarzinom ist die häufigste endokrine Neoplasie mit einer Inzidenz von 4/100000/Jahr (die Inzidenz hat insbesondere in der letzten Dekade weltweit deutlich zugenommen). Der Manifestationsgipfel liegt jenseits des 50. Lebensjahrs.
→ II: Bei der differenzierten Form sind Frauen 3 x häufiger als Männer betroffen.
→ 1) Follikuläres SD-Karzinom: (35% der Fälle) Vermehrtes Auftreten in Jodmangelgebieten mit hoher Struma-Inzidenz. Die Häufigkeit nimmt mit dem Alter deutlich zu.
→ 2) Papilläres SD-Karzinom: (50% der Fälle) Häufig in Gebieten mit normaler Jodversorgung. Kann in jedem Alter auftreten; betrifft vermehrt jüngere Patienten
→ III: Beim anaplastischen - und medullären Schilddrüsenkarzinom ist die Geschlechterverteilung gleich.
→ Ätiologie: Die Pathogenese der differenzierten Schilddrüsenkarzinome ist nicht genau bekannt, jedoch erhöhen ionisierende Strahlungen das Erkrankungsrisiko drastisch. Risikofaktoren für die Entwicklung einer Maglinität aus einem Knoten sind nach der American-Association of Clinical Endocrinologists (= AACE) u.a. Bestrahlung des Kopfes oder Kehlkopfbestrahlung in der Vorgeschichte, positive Familienanamnese mit einem medullären Schilddrüsenkarzinom, eine MEN 2, ein fixierter derber Knoten, der ein schnelles Wachstum aufweist, männliches Geschlecht sowie Alter < 20. Lebensjahr oder > 70. Lebensjahr und nicht zuletzt Symptome wie vergrößerte Lymphknoten, Heiserkeit, Schmerzen, Dysphagie und Dyspnoe, etc. Weitere prädisponierende Faktoren sind insbesondere genetische Syndrome wie die familiäre Polyposis, das Cowden-Syndrom, Werner-Syndrom, etc.
→ Pathogenese: Im Vordergrund stehten zahlreiche molekulargenetische Veränderungen im Bereich der Onkogene und Tumorsupressorgene:
→ I: Follikuläres Schilddrüsenkarzinom: Sowohl beim follikulären Adenom als auch beim Karzinom lassen sich in 20-30% der Fälle Mutationen in der ras-Protoonkogenfamilie (H-ras, K-ras und N-ras) eruieren. Es wird in diesem Zusammenhang angenommen, dass das follikuläre Adenom ein frühes Phänomen der Tumorgenese darstellt.
→ II: Papilläres Schilddrüsenkarzinom: Im Mittelpunkt der Pathogenese steht die Aktivierung RET-PTC-Onkogens aufgrund einer Translokation des RET-Protoonkogens und des H4-Gens. Weitere molekulare Veränderungen des papillären Schilddrüsenkarzinoms wie trk-Rearrangement, Überexpression von ras-Onkogen-Produkten, etc. sind weniger spezifisch, weisen jedoch auf einen besonders aggressiven Charakter und eine rapide Tumorprogression hin.
→ Pathohistologie:
→ I: Papilläres Schilddrüsenkarzinom: Pathomorphologisch lassen sich beim papillären Schilddrüsenkarzinom verschiedene Varianten unterscheiden:
→ 1) Papilläres Mikrokarzinom: Nach der WHO werden alle papillären SD-Karzinome, deren maximaler Durchmesser < 1cm ist, als Mirkokarzinome bezeichnet, unabhängig davon, ob der Tumor gekapselt ist oder nicht.
→ 2) Gekapseltes papilläres Karzinom: Dieser Karzinomtyp macht etwa 10% aller papillären Schilddrüsenkarzinome aus und wird komplett von einer Faserkapsel ummantelt.
→ 3) Follikuläre Variante: Des papillären Schilddrüsenkarzinom. Diese Form weist ein fast reines follikuläres Wachtumsmuster auf, und es bedarf beim Nachweis papillärer Strukturen (charakteristische Kernveränderungen sowie bevorzugte lymphogene Metastasierung) größter Sorgfalt.
→ 4) Weitere Typen: Weitere pathomorphologische Varianten des papillären Schilddrüsenkarzinoms sind u.a.:

→ II: Follikuläres Schilddrüsenkarzinom: Diese maligne Neoplasie hat ihren Ursprung im Follikelepithel der Schilddrüse und besitzt pathomorphologisch auch verschiedene Varianten, die sich z.T. in ihrer Prognose stark differenzieren.

→ Klassifikation: Pathohistologische Einteilung der Schilddrüsenkarzinome:
→ I: Differenziertes SD-Ca:
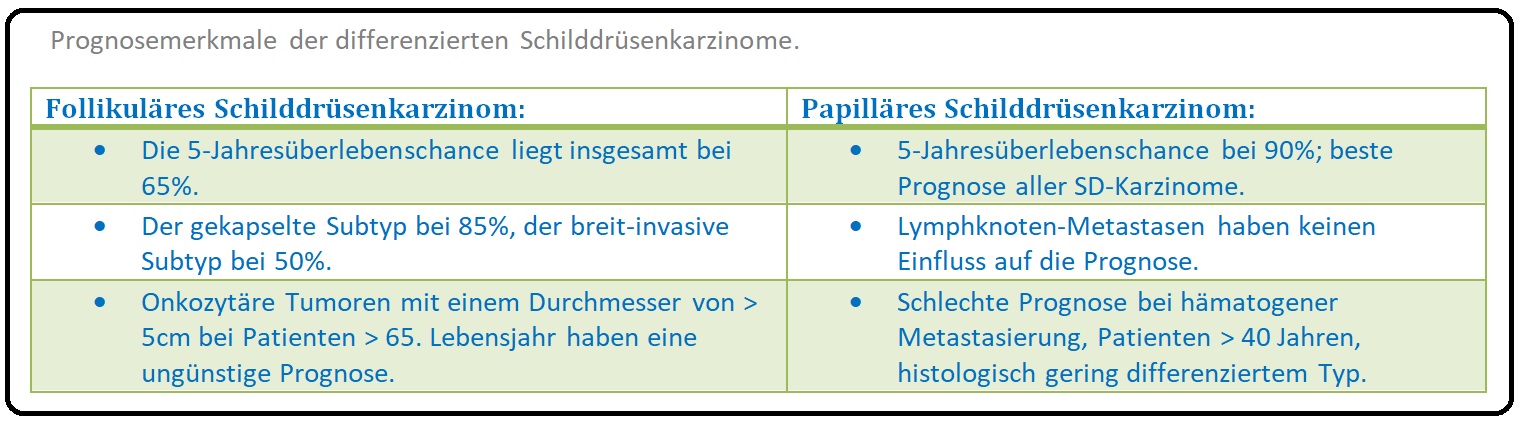
→ 1) Follikuläres SD-Ca: Das follikuläre SD-Karzinom macht 35% aller Malignome der SD aus und stellt somit die 2.-häufigste Karzinomart dar; es handelt sich zumeist um ein solitäres Karzinom und metastasiert frühzeitig hämatogen in Knochen und Lunge.
→ 2) Papilläres SD-Ca: Ist mit 50% das häufigste SD-Karzinom und metastasiert vorwiegend lymphogen in die regionären Lymphknoten, z.T. bevor der Primärtumor nachweisbar ist (Mikrokarzinom < 1cm, das lange Zeit asymptomatisch bleibt). Der Tumor tritt überwiegend multifokal auf und lässt sich gut mit einer 131Jod-Therapie behandeln.

→ II: Undifferenziertes SD-Ca: Das anaplastisches Schilddrüsenkarzinom stellt mit 5% der Fälle eine sehr seltene Erkrankung dar. Zudem nimmt es nicht am Jodumsatz teil, sodass eine 131Jod-Therapie ungeeignet ist.
→ III: C-Zell-Karzinom: (5%) Ist eine von den medullären C-Zellen ausgehende Neoplasie, die vermehrt Calcitonin produziert (Rezidivindikator).
→ Klinik: Lange Zeit asymptomatisch; erst im fortgeschrittenen Stadium manifestiert sich eine Symptomatik aufgrund eines infiltrativen Wachstums in die Nachbarorgane wie Trachea, Ösophagus, sowie Nervenbeteiligung. Klinische Zeichen sind u.a.:
→ I: Lokale Zeichen: Derber, höckriger Knoten, fixierte Haut, geschwollene nicht-schmerzhafte Lk im zervikalen und supraklavikulären Bereich.
→ II: Globusgefühl und Dysphagie,
→ III: Heiserkeit durch eine Recurrensparese,
→ IV: Inspiratorischer Stridor und Dyspnoe bei Befall der Trachea.
→ V: Horner-Syndrom: Selten, mit Miosis, Ptosis und Endophthalmus.
→ VI: Fernmetastasen betreffen besonders die Lunge und das Skelettsystem.
→ Diagnose:
→ I: Die Diagnose wird häufig bei der Abklärung einer nodulären Struma gestellt.
→ II: Bestimmung des Thyreoglobulins (Tumormarker bei Rezidivabklärung, evtl. auch TPA = tissue-polypeptid-antigen)
→ III: Sonographie: Besonders verdächtig sind unregelmäßig begrenzte, echoarme Areale mit Mikrokalzifikationen. Weitere sonographische Malignitätsbefunde sind u.a.:
→ 1) Kein Halo,
→ 2) Intranoduläre Vaskularisierung etc.
→ IV: Szintigraphie: Kalte Knoten, die nicht speichern.
→ V: Fernmetastasen: Zum Ausschluss Röntgen-Thorax, CT, Sono-Abdomen, Knochen-Szintigraphie.

→ Klinisch-relevant: Ein szintigraphisch kalter SD-Knoten, der sonographisch nicht echofrei ist, ist immer tumorverdächtig. Folgende Untersuchungen sind hierbei obligat.
→ A) Feinnadelbiospie mit Aspirationszytologie ist in 90% wegweisend.
→ B) Bei negativem Befund und fortbestehendem Tumorverdacht; operative Tumorentfernung und nachfolgende histologische Untersuchung.
→ Differenzialdiagnose: Vom differenzierten Schilddrüsenkarzinom sind u.a. nachfolgende Erkrankungen abzugrenzen:
→ I: Zystische Schilddrüsenveränderungen: Einschließlich der Zystenblutungen.
→ II: Thyreoditis: Wie die Hashimoto-Thyreoiditis oder die subakute Thyreoiditis der Quervain.
→ III: Struma nodosa: (euthyreote Struma) Mit benignen, regressiven Knoten; die Abgrenzung ist z.T. sehr schwierig, sodass eine Thyreoidektomie erfolgen muss.
→ IV: Weitere Differenzialdiagnosen: Sind insbesondere:
→ 1) Lymphom der Schilddrüse sowie
→ 2) Metastasen extrathyreoidaler Malignome.
→ Therapie: Die genaue histologische Bestimmung des differenzierten Schilddrüsenkarzinoms für die Behandlung von besonderer Bedeutung.
→ I: Operative Therapie:
→ 1) In den meisten Fällen ist eine radikale totale Thyreoidektomie mit Entfernung der Lymphknoten im zentralen Kompartiment indiziert. Der gleichzeitige, metastatische Befall der regionären Lymphknoten führt zu einer zusätzlichen chirurgischen Neck-Dissektion.
→ 2) Bei einem solitären, papillären Mikrokarzinom ohne Nachweis von Lymphknotenmetastasen kann die Hemithyreoidektomie eine kurative Therapie sein.
→ II: Ablative Radiojodtherapie: In der 3.-4. postoperativen Woche erfolgt ein 131Jod-Ganzkörperscan zum Ausschluss/Nachweis von Thyreozytenresten und Metastasen; sind Metastasen eruierbar, sollte eine hochdosierte Radiojodtherapie in mehreren Fraktionen, bis kein Jod-speicherndes Gewebe mehr nachweisbar ist, durchgeführt werden.
→ III: Weitere Therapiemaßnahmen:
→ 1) Im Anschluss (Radiojodtherapie) ist eine hochdosierte Hormonlangzeittherapie mit Levothyroxin (150-300µg/d) zur Suppression von TSH, (um die wachstumsfördernde Wirkung von TSH auf evtl. Metastasen zu unterbinden) indiziert.
→ 2) Evtl. externe Radiatio als pallitative Therapie zur Reduktion von Tumorkomplikationen z.B. bei vehementen Schmerzen.
→ IV: Nachsorge: Kontrolluntersuchungen im Abstand von 6 Monaten sind obligat mit:
→ 1) Bestimmung des Thyreoglobulins (TG). Dieses wird vom normalen SD-Gewebe sowie von den Zellen des SD-Karzinoms gebildet. Durch die radikale Thyreoidektomie spricht ein Anstieg des TG für ein Tumorrezidiv bzw. Metastasen.
→ 2) Evtl. Thyreogobulin-Bestimmung nach Gabe von rekombinantem humanem TSH (rhTSH) zum Nachweis von Tumorrezidiven/Metastasen.
→ 3) Bei Verdacht auf ein Rezidiv bzw. eine Metastase sollte immer ein 131Jod-Ganzkörperscan durchgeführt werden.
→ 4) Weitere Nachweisoptionen von Metastasen sind Röntgen-Thorax, CT, Ganzkörperskelett-Szintigraphie etc.
→ Prognose: Bei adäquater Therapie liegt die 5-Jahresüberlebenschance beim papillären Schilddrüsenkarzinom bei > 90% und hat von allen SD-Karzinomen die beste Prognose. Beim follikulären SD-Karzinom liegt sie bei 65%. Weitere wichtige Prognosefaktoren sind u.a. Tumorgrößer, histologischer Typ, Ausmaß der extrathyreoidalen Invasion, Lymphknoten- und Fernmetastasen sowie das Lebensalter.
- Details
- Kategorie: Schilddrüse und Nebenschilddrüse
- Zugriffe: 17480
→ Definition:
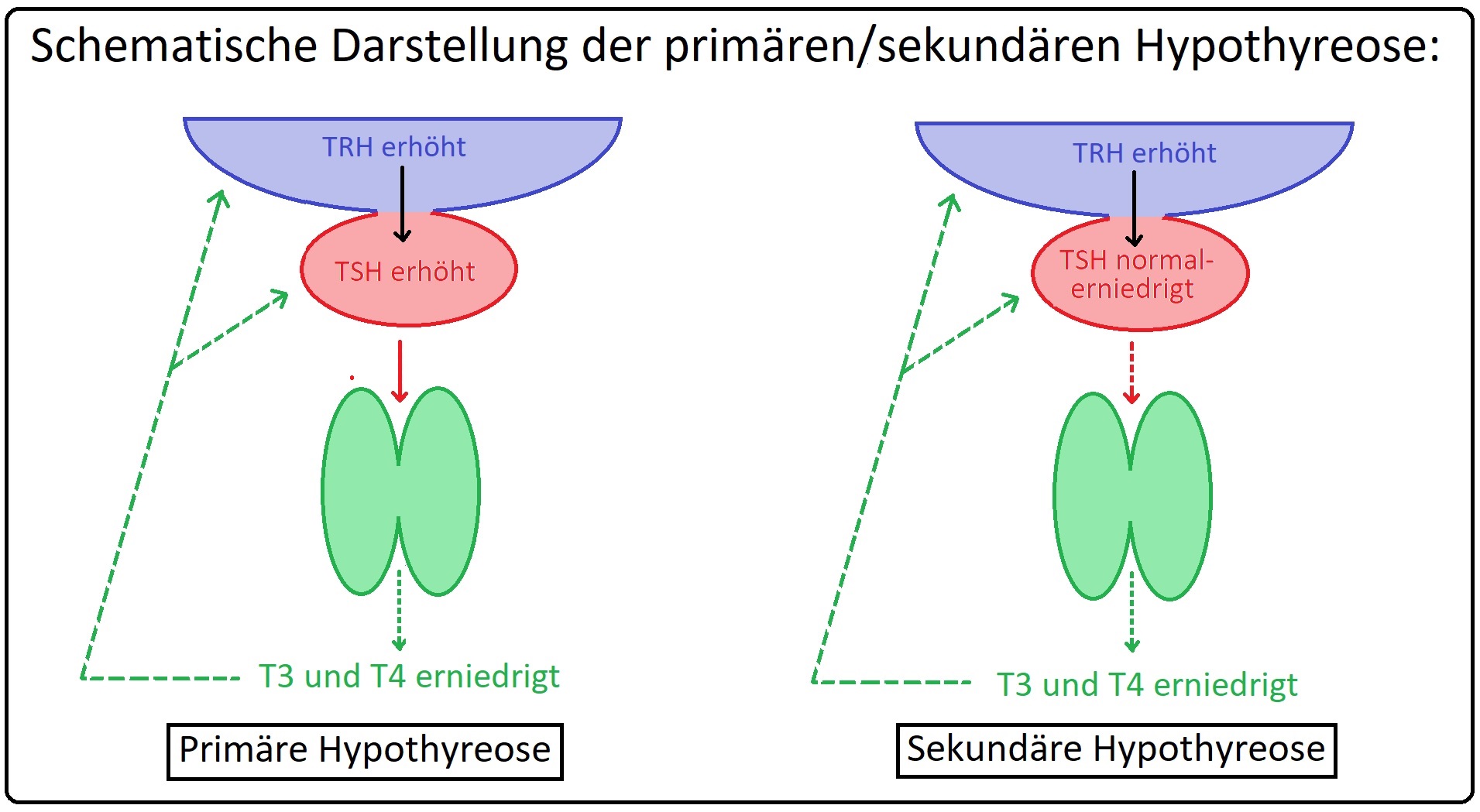
→ I: Sekundäre Hypothyreose: Bei dieser Form manifestiert sich eine Störung der thyreotropen Zellen des Hypophysenvorderlappens im Rahmen einer Hypophysenvorderlappeninsuffizienz bei intakter Schilddrüse. Zudem existiert ein familiär auftretender TSH-Mangel, der durch das Fehlen des Transkriptionsfaktors PIT-1 oder dessen Vorläufers PROP-1 verursacht wird.
→ II: Tertiäre Hypothyreose: Es handelt sich um eine hypothalamisch-bedingte Hypothyreose mit Insuffizienz des Hypothalamus und konsekutivem TRH-Mangel.
→ Epidemiologie: Die sekundäre Hypothyreose ist selten; die tertiäre stellt eine Rarität dar.
→ Ätiopathogenese:
→ I: Sekundäre Hypothyreose:
→ 1) Ursachen sind vor allem:
→ A) Verdrängung der TSH-produzierenden Zellen durch einen Hypophysentumor (meist Adenome).
→ B) Zustand nach Hypophysektomie und Hypophysenbestrahlung, sowie
→ C) Zustand nach Schädel-Hirn-Traumata.
→ 2) Charakteristischerweise fehlt bei intaktem Hypothalamus die TSH-Produktion der Hypophyse. In der Folge manifestiert sich ein TRH-Anstieg, der mit einem Prolaktinanstieg einhergeht.
→ II: Tertiäre Hypothyreose: Sehr seltene Hypothyreoseform bedingt u.a. durch z.B. tumoröse Prozesse oder einen kongenitalen Anlagedefekt der Kerngebiete der TRH-Produktion. Folge ist eine Erniedrigung der TSH- und der Schilddrüsenhormonproduktion.
→ Klinik:
→ I: Die klinischen Symptome gleichen denen der primären Hypothyreose, nur entwickeln sie sich meist langsamer und erreichen das Symptom-Ausmaß nicht.
→ II: Evtl. können sich klinisch im Zuge der Hypophysenvorderlappeninsuffizienz weitere Hormonproduktionsstörungen (z.B. ACTH) herauskristallisieren.
→ III: Symptome der Hypothyreose sind:
→ 1) Psychomotorisch: Müdigkeit, Somnolenz, psychomotorische Verlangsamung, Muskelschwäche.
→ 2) Vegetativ: Hypothermie, Kälteintoleranz, Hypotonie (mit diastolischer Hypertonie), Bradykardie, Obstipation und Hypoventilation.
→ 3) Psychische Störungen: Geistige Retardierung, Gedächtnisstörungen, depressive Verstimmung.
→ 4) Weitere Symptme: Sind Gewichtszunahme, Gesichtsödem (periokulär), Myxödem (durch Einlagerung von Glykosaminoglykane hervorgerufenes, nicht-eindrückbares Ödem), schuppige, teigige, bleich-gelbliche Haut, trockene Haare, evtl. Haarverlust, Menstruationsstörungen, Infertilität, erhöhte Abortrate etc.
→ Komplikationen:
→ I: Sehr selten isoliertes Myxödemkoma,
→ II: Häufiger hypophysäres Koma bei Hypophysenvorderlappeninsuffizienz.
→ Diagnose: Die Diagnose wird klinisch bzw. laborchemisch gestellt.
→ I: Labor:
→ 1) TSH erniedrigt (untere Normbereich); fT3/fT4 auch erniedrigt.
→ 2) Thyreotroper Funktionstest: Bei der sekundären Hypothyreose findet nach Gabe von TRH kein ausreichender (fehlender) Anstieg von TSH und der SD-Hormone statt.
→ II: CT/MRT: Zur Tumorsuche bzw. zum Ausschluss eines Empty-Sella-Syndroms (= Es handelt sich um eine Erweiterung des Subarachnoidalraumes infolge einer hernienartigen, liquorgefüllten Aussackung, die in die Sella turcica hineinreicht. In der Folge kann sich durch Kompression eine HVL Insuffizienz entwickeln).
→ Differenzialdiagnose: Von der sekundären - bzw. tertiären Hypothyreose müssen insbesondere nachfolgende Erkrankungen abgegrenzt werden:
→ I: Depression,
→ II: Demenz,
→ III: Herzinsuffizienz anderer Genese.
→ Therapie:
→ I: Die Behandlung erfolgt wie bei der primären Hypothyreose mit L-Thyroxin.
→ II: Gerade bei älteren Patienten ist, aufgrund der kardiovaskulären Komplikationen (wie Herzrhythmusstörungen, KHK und Myokardinfarkt), eine einschleichende Dosierung von 12,5-25µg/d morgens und anschließend eine schrittweise Steigerung alle 1-2 Wochen auf eine Erhaltungsdosis von 75-150µg/d indiziert.
→ Klinisch-relevant:
→ A) Vor einer L-Thyroxin-Therapie der sekundären Hypothyreose ist der Ausschluss einer Nebenniereninsuffizienz obligat.
→ B) Da TSH als Kontrollparameter für die medikamentöse Einstellung nicht herangezogen werden kann, orientiert sich Behandlung an der fT4-Konzentration (sollte im mittleren Normbereich liegen).
→ Prognose: Sie richtet sich nach der Grunderkrankung. Bei frühzeitiger adäquater Therapie sollten Spätkomplikationen wie Arteriosklerose und Herzinsuffizienz vermieden werden.