→ Definition: Bei der Myasthenia gravis (= neuromuskuläre Erkrankung) handelt es sich um eine autoimmunologische Störung der neuromuskulären Erregungsübertragung mit Antikörper-Bildung. Zirkulierende polyklonale IgG-Antikörper führen primär zu einer Blockade, sekundär zu einer Abnahme der postsynaptischen nikotinergen Acetylcholin-Rezeptoren an der motorischen Endplatte. Leitsymptom ist eine belastungsabhängige Muskelschwäche ohne Myalgie.
→ Epidemiologie:
→ I: Die Myasthenia gravis stellt mit einer Prävalenz von 5-10/100000 Einwohnern eine seltene Erkrankung dar.
→ II: Man unterscheidet nach dem Manifestationszeitpunkt insbesondere zwischen 2 Formen:
→ 1) Juvenile Form: Hierbei tritt die Erkrankung vor dem 16. Lebensjahr auf.
→ 2) Adulte Form: Ein Erkrankungsgipfel bei Frauen liegt zwischen der 2.-3. Lebensdekade, bei Männern jenseits der 5. Lebensdekade (50.-70. Lebensjahr).
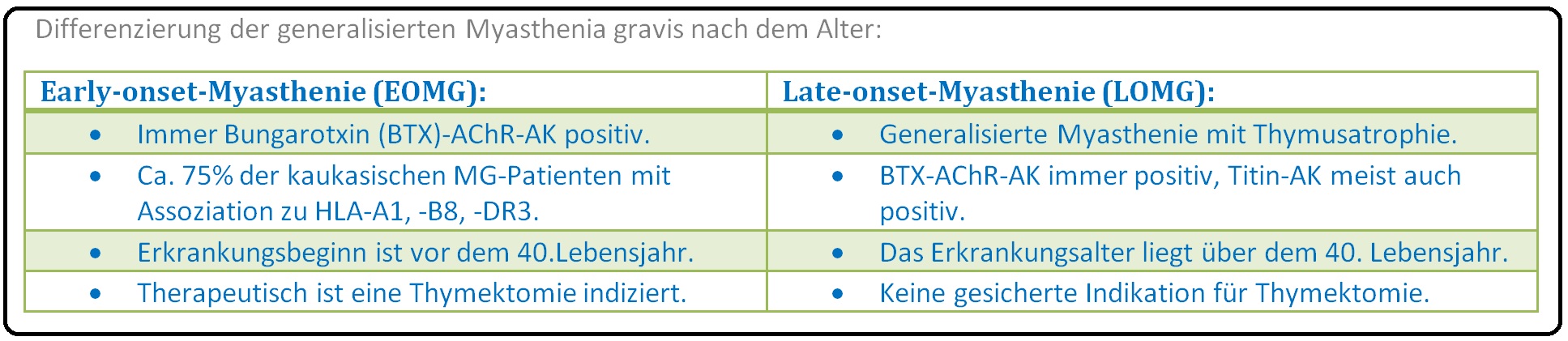
→ III: In nur 3% der Fälle manifestiert sich eine familiäre Häufung insbesondere bei Assoziation mit HLA-B8 und DR3.
→ Ätiologie: Es bestehen verschiedene Faktoren:
→ I: Genetische Disposition: In 3-5% der Fälle manifestiert sich eine genetische Disposition; hierfür spricht auch eine Assoziation zu HLA-B8 und DR3.
→ II: Thymusveränderungen: In bis zu 65% der Fälle ist eine Thymushyperplasie eruierbar. In größerer Regelmäßigkeit lassen sich im Thymus ortsfremde Myoidzellen nachweisen, die Acetylcholin-Rezeptoren tragen. Man nimmt an, dass die Acetylcholin-Rezeptoren von den im Thymus gebildeten T-Lymphozyten als (Auto-)-Antigene erkannt werden. Diese aktivierten T-Zellen wandern in die Peripherie aus und induzieren immunkompetente B-Lymphozyten zur Produktion von Antikörpern gegen Ach-Rezeptoren, die im Bereich der Postsynapse an Acetylcholin-Rezeptoren binden.
→ III: Paraneoplastisch: 10-15% der Patienten mit Myasthenia gravis weisen ein Thymom auf. Anti-Titin-Antikörper bei jüngeren Patienten (< 50. Lebensjahr) sind häufig mit einem Thymom assoziiert.
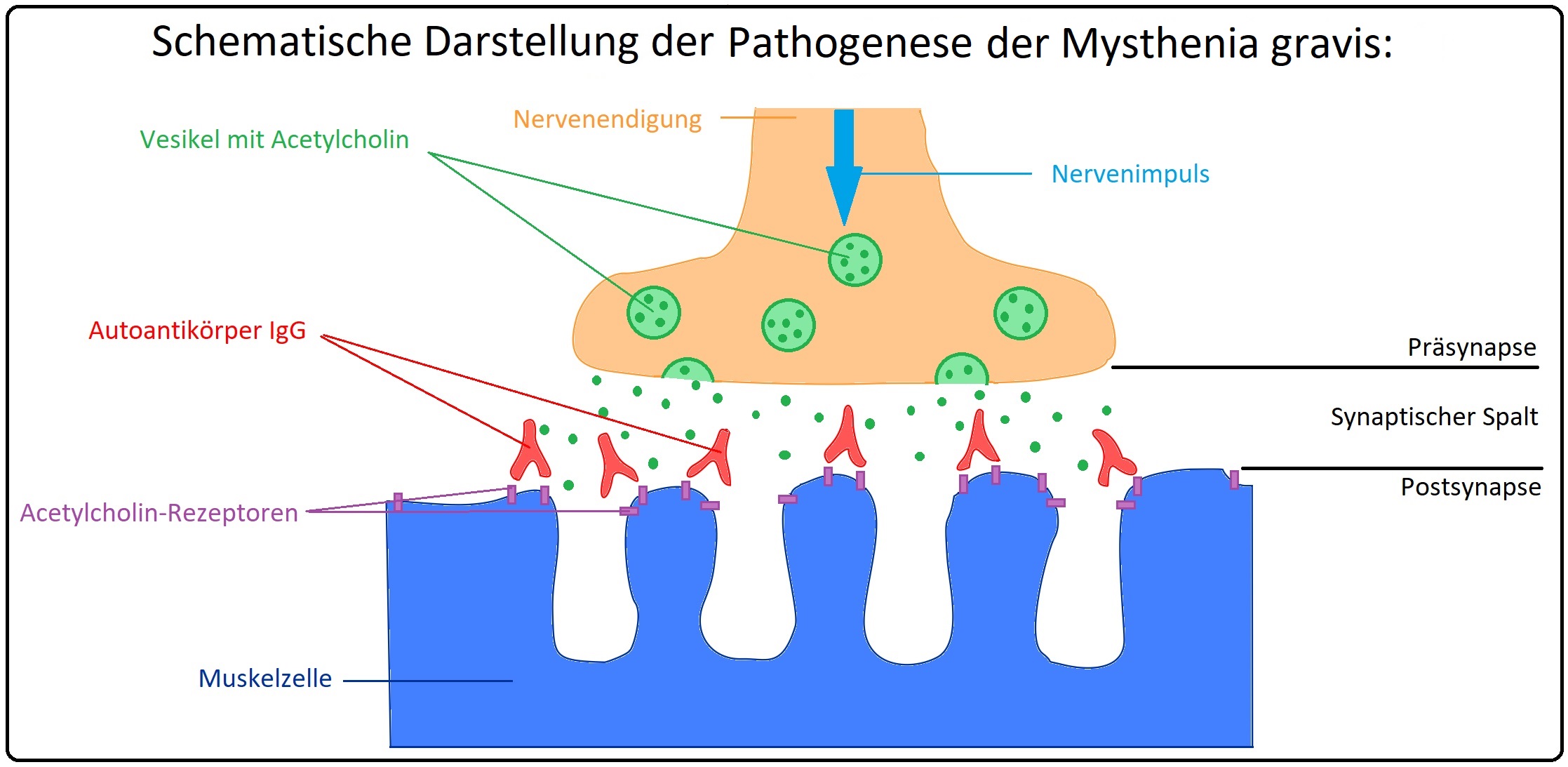
→ Pathogenese:
→ I: Pathogenetisch handelt es sich um eine Autoimmunerkrankung mit Bildung von Autoantikörpern vom IgG-Typ gegen postsynaptische nikotinerge Aetylcholin-Rezeptoren der Muskulatur. Diese polyklonalen Antikörper binden an
postsynaptische ACh-Rezeptoren und induzieren durch Blockade eine Abnahme der verfügbaren Rezeptoren an der motorischen Endplatte. Durch Aktivierung des Komplement-Systems kommt es zur:
→ 1) Zerstörung postsynaptischer Membranen durch den „Membrane-attack-Complex“ und
→ 2) Rezeptoraggregation mit konsekutiv beschleunigtem Abbau von Rezeptoren.
→ II: Wichtige Autoantikörper bei der Myasthenia gravis sind insbesondere:
→ 1) AChR-Antikörper (lassen sich in 90% der Fälle nachweisen.
→ 2) MuSK-Antikörper: Stelle Antikörper gegen muskelspezifische Rezeptor-Tyrosinkinase dar und stehen zumeist für dramatischen Krankheitsverlauf mit häufigen myasthenen Krisen.
→ 3) LPR4-Antikörper: Nachweis von Antikörper gegen Lipoprotein-related-Protein 4 bei anti-AChR und anti-MuSK Sero-Negativität (Verkleinerung der serologischen Lücke).
→ 4) Anti-Titin-AK: Titin ist ein wichtiges Strukturprotein des Sakrolemms. Titin-Antikörper, insbesondere gegen das Filament MGT-30, sind häufig bei Patienten unter dem 50. Lebensjahr mit einem Thymom vergesellschaftet.

→ III: Häufige Tiggermechanismen für die Auslösung einer myasthenen Reaktion sind u.a.:
→ 1) Menstruation, Insolation und Infektionen sowie während der Schwangerschaft.
→ 2) Psychogene Stressoren,
→ 3) Medikamenten-induziert: Durch z.B. Chinin, Chloroquin, Penicillamin, Antibiotika (z.B. Aminoglykoside, Makrolide, Gyrasehemmer, Trtracycline, etc), Benzodiazepine, Antidepressiva vom Amitriptylin-Typ, Lithium, Antipsychotika (Chlorpromazin, Promazin), Cholesterinsenker (Statine) und nicht zuletzt Chinidin, Ajmalin, Beta-Blocker, Procainamid sowie Kalzium-Antagonisten, etc.
→ Klinisch-relevant: Die Myasthenia gravis ist vermehrt mit nachfolgenden (Autoimmun-) Erkrankungen vergesellschaftet; hierzu zählen:
→ A) Hyper- und Hypothyreose, endokrine Orbitopathie und die Thyreotoxikose.
→ B) Kollagenosen wie der SLE, Sjögren-Syndrom, Dermatomyositis, etc.
→ C) Weitere Erkrankungen: Wie die rheumatoide Arthritis, Sarkoidose, Morbus Crohn und Colitis ulcerosa, etc.
→ Klassifikation: Die Myasthenia gravis kann nach verschiedenen Gesichtspunkten unterteilt werden:
→ I: Eine Klassifikation berücksichtigt das Alter des Patienten beim Auftreten der klinischen Symptomatik und unterscheidet zwischen einer juvenilen - (im Kindes- und Jugendalter auftretend) und einer adulten Form.
→ II: Einteilung der MG nach dem Schweregrad der Erkrankung nach Ossermann:

→ III: Weitere Formen: Der Myasthenie sind u.a.:
→ 1) Neonatale Myasthenie: Bei etwa 10% der Neugeborenen myasthener Mütter erfolgt eine passive Übertragung der Antikörper diaplazentar oder mit dem Kolostrum. Hierbei kommt es zur Spontanheilung, sodass die Therapie nur symptomatisch existiert.
→ 2) Kongenitale myathene Syndrome: Stellt eine sehr seltene Gruppe von genetisch-bedingten Störungen der prä- und postsynaptischen Strukturen. Es handelt sich zumeist um eine rezessiv vererbte Erkrankung; Neumutationen existieren aber auch. Eine frühzeitige Diagnosestellung ist bei dieser Form von besonderer Bedeutung, da ansonsten ein hohes Mortalitätsrisiko z.B. im Rahmen von Infekten besteht (sudden infant Death).
→ Klinik: Bei der Myasthenia gravis muss zwischen einer rein okulären Form, bei der definitionsgemäß nur die äußeren Augenmuskeln betroffen sind, und den generalisierten Formen unterschieden werden.
→ I: Initialstadium: Zumeist zeigen sich okuläre Beschwerden wie Doppelbilder (frühzeitig beim Seitwärtsblicken), die im Tagesverlauf oder unter Anstrengung zunehmen, und eine ein- bzw. beidseitige Ptosis. Nur bei 10-20% der Patienten bleibt die Myasthenie innerhalb der ersten 2 Jahren auf die Augen beschränkt. Bei der MuSK-AK positiven Myasthenie zeigt sich nur sehr selten eine okuläre Symptomatik.
→ II: Bulbäre Symptome: (Fazio-pharyngeal) Mit schlaffen Gesichtszügen (= Facies myopathica), Dysarthrie (mit näselnder kloßiger schlecht artikulierter Stimme), Kauschwäche (insbesondere aufgrund des M. masseter) und Dysphagie. Die Symptome nehmen im Verlauf der Mahlzeit zu. In diesem Zusammenhang kommt es häufig zu typischen Gewichtsabnahme, die häufig übersehen wird.
→ III: Generalisierte Form:
→ 1) Muskelschwäche, die insbesondere proximal betont und belastungsabhängig ist; gelegentlich zeigt sich ein asymmetrischer Befall.
→ 2) Die Muskelschwäche nimmt typischerweise im Tagesverlauf zu und es zeigt sich ein Beschwerderückgang in Ruhephasen.
→ 3) Variabler Krankheitsverlauf mit Fluktuation der Muskelschwäche über Monate bis Jahre. Eine akute Exazerbation erfolgt u.a. durch Allgemeininfekte.
→ 4) Bei der MuSK-AK-positiven Form wird frühzeitig eine Muskelatrophie sichtbar.
→ 5) Typische kardiale Manifestationen der Myasthenie sind unspezifische EKG-Veränderungen, Herzrhythmusstörungen sowie Herzinsuffizienz.
→ Komplikationen: Wichtige und z.T. schwerwiegende Komplikationen der Myasthenia gravis (MG) sind u.a.:
→ I: Bulbäre Paresen mit Schluckstörungen und Gefahr der Aspiration (Pneumonie bzw. Aspirationspneumonie).
→ II: Myasthene Krisen sowie
→ III: Beteiligung der Atemmuskulatur mit der Gefahr der Ateminsuffizienz bis hin zum Tod.
→ Diagnose:
→ I: Anamnese: Beginn der Beschwerden und Tagesverlauf sowie Veränderung der Symptomatik unter Belastung.
→ II: Klinische Untersuchung:
→ 1) Durch repetitives Öffnen oder Schließen der Hand oder Reklination und Neigung des Kopfes tritt schnell eine Ermüdung der Muskulatur auf.
→ 2) Simpson-Test: Anhaltender Aufwärtsblick (> 1 min.) führt zu einer Zunahme einer Ptosis.
→ 3) Tensilon-Test.

→ III: Labor:
→ 1) Mit Bestimmung von Schilddrüsenhormonen, Blutzuckertagesprofil und Nierenparametern.
→ 2) Antikörperbestimmung insbesondere von Acetylcholin-Rezeptor-AK, AK gegen muskelspezifische Tyrosinkinase (häufig bei Patienten mit bulbopharyngeale Manifestation, Anti-Titin-AK (Marker für eine paraneoplastische Myasthenie bei Thymom).
→ 3) Screening von weiteren Autoimmunerkrankungen wie SLE, rheumatoider Arthritis, Morbus Bechterew, entzündliche Darmerkrankungen, Glomerulonephritis, etc.
→ III: EMG: (Mit repetitiver Reizung 3 Hz über 2 sec.).
→ 1) Reizorte sind bei diesem Untersuchungsverfahren:
→ A) N. accessorius am Hinterrand des M. sternocleidomastoideus).
→ B) N. axillaris am Erb-Punkt (Ableitung von M. deloideus).
→ C) N. fascialis am Foramen stylomastoideus (Ableitung vom M. orbicularis oculi).
→ 2) Abnahme der Amplitude des motorischen Summenpotenzials des 5. Reizes im Vergleich zum 1. Reiz.
→ 3) Ein Flächendekrement von > als 10% oder ein Amplitudendekrement > 12-15%.
→ 4) Verschwinden des Amplitudendekrements nach Gabe eines Cholinesterase-Hemmers.
→ IV: Bildgebung:
→ 1) CCT/cMRT: Insbesondere bei der okulären Myasthenie zum Ausschluss von Raumforderungen.
→ 2) MRT: (Spiral CT) des Thorax mit Kontrastmittel zur Eruierung eines möglichen Thymoms bzw. einer Malignität.
→ 3) PET: (mit Fluordesoxyglukose) Nachweis versprengten Thymomresten.
→ Differenzialdiagnose: Von der Myasthenia gravis müssen insbesondere nachfolgende Erkrankungen abgegrenzt werden:
→ I: Die okuläre Myositis oder Polyneuropathie cranialis sind nicht belastungsabhängig und sprechen auch nicht auf die Tensilon-Applikation an.
→ II: Auch die chronisch progressive Ophthalmoplegie (von Graefe) ist mit dem Untersuchungsverfahren abzugrenzen.
→ III: Kearns-Syre-Syndrom: Bei diesem Syndrom neben der Ophthalmoplegia noch weitere somatische Pathologien wie u.a. eine Kardiomyopathie.
→ IV: Lambert-Eaton-Syndrom,
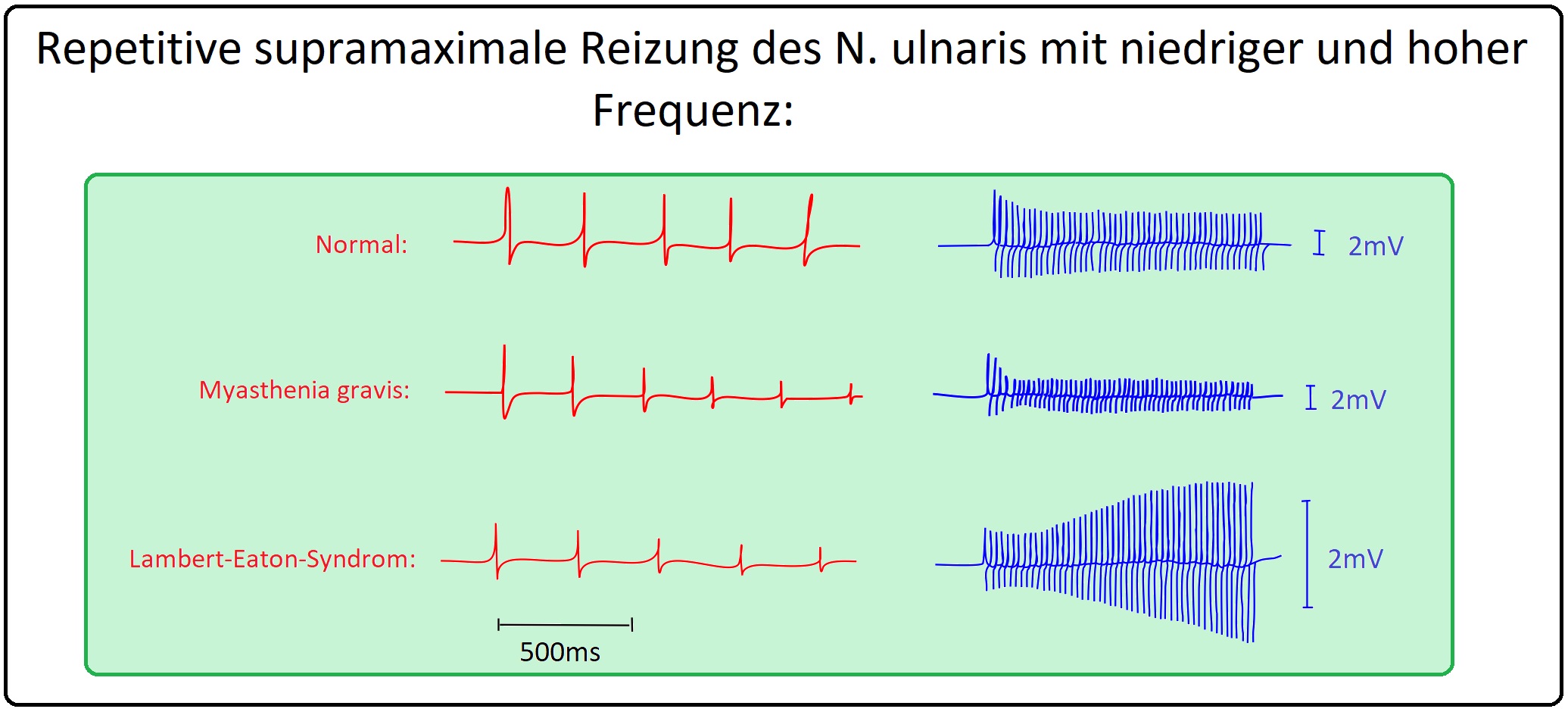
→ V: Mitochondriopathien.
→ VI: Weitere Erkrankungen: Wie insbesondere:
→ 1) Glykogenose Typ V,
→ 2) Bulbärparalyse,
→ 3) Botulismus,
→ 4) Schilddrüsendysfunktion,
→ 5) Dissoziative Störungen (Ausschlussdiagnose), etc.
→ Therapie: Die Therapie der Myasthenia gravis richtet sich insbesondere nach dem Schweregrad der Erkrankung und dem Antikörpertiter. Allgemeinmaßnahmen sind z.B. regelmäßige Erholungspausen, keine massive körperliche Belastung, Vermeiden
von fieberhaften Infekten oder Lebensimpfstoffen, etc. Die wichtigsten Therapieoptionen sind:
→ I: Konservative Therapie: Ziel ist die Remission der klinischen Symptomatik sowie die Wiederherstellung der Arbeitsfähigkeit bei geringen bzw. fehlenden Nebenwirkungen durch die Immunsuppression.
→ 1) Pyridostigmin: Hierbei handelt es sich um einen Cholinesterase-Hemmer, der in einer Dosis von 30-60mg/alle 4-5 Stunden (bis zu einer Maximaldosis von 360mg/d) über den Tag oral appliziert wird. Es zeigt sich ein besonders geringes Ansprechen bei der MuSK-positiven-Myasthenie.
→ 2) Kortikosteroide: Wie Methylprednisolon initial in einer Dosis von 15-20mg/d einschleichend steigernd um 5mg/Wochen bis zu einer Zieldosis von 0,5-1,5mg/kgKG/d. Die einschleichende Therapiedosierung soll einer möglichen Symptomverschlechterung entgegenwirken.
→ 3) Weitere Immunsupressiva: Azathioprin gelten als Standardtherapie bei mittelschwerer bis schwerer Myasthenie; zu Beginn ausschließlich in Kombination mit Kortison. Bei Unverträglichkeit kann Ciclosporin A alternativ Mycophenolat oder Tacrolimus verabreicht werden.
→ II: Operative Therapie: Es erfolgt eine transternale Thymektomie nach sorgfältiger Exploration des Mediastinums auf ektopes Thymusgewebe (bei Thymomen < 2cm wird die Operation zunehmend thorakoskopisch durchgeführt). Wichtige Indikationen sind u.a.:
→ 1) Thymom-Nachweis in jedem Alter.
→ 2) Bei ACh-Rezeptor-AK positiver Myasthenie im Alter zwischen 15-50 Jahren und kurzer Krankheitsdauer (< 2 Jahre).
→ 3) Unzureichender Effekt der medikamentösen Therapie.
→ Prognose:
→ I: Abgesehen von den foudroyanten Krankheitsverläufen ist die Gesamtprognose der Myasthenia graivs mit der Einführung der immunsuppressiven Langzeittherapie entscheidend verbessert worden (weist heutzutage zumeist einen langsam progredienten Krankheitsverlauf auf).
→ II: Häufig manifestiert sich bei den Patienten im Verlauf eine Generalisierung; die rein okuläre Form besteht nur selten und hat aber eine sehr gute Prognose (ohne Minderung der Lebenserwartung).
→ III: Prognostisch ungünstig sind u.a.:
→ 1) Hohes Lebensalter,
→ 2) Schwere generalisierte Form der Myasthenie und nicht zuletzt
→ 3) Neoplastische Thymusveränderungen.