→ Definition: Bei der Sarkoidose handelt es sich um eine Systemerkrankung unklarer Genese mit Ausbildung von nichtverkäsenden, epitheloidzelligen Granulomen (mit Riesenzellen, ohne zentrale Nekrosebildung) vorwiegend im Bereich der thorakalen Lymphknoten und der Lunge, aber auch in anderen Organen (z.B. Leber, Milz, aber auch die Haut mit Erythema nodosum oder die Augen mit einer Iridozyklitis, etc.).
→ Epidemiologie:
→ I: Die Sarkoidose stellt die häufigste interstitielle Lungenerkrankung dar und ihre Prävalenz liegt bei 40/100000 Einwohnern in Deutschland.
→ II: Die Erkrankung weist ein Nord-Süd Gefälle (nach Süden hin abnehmend) auf mit einem gehäuften Auftreten in Schweden und Island.
→ III: Die Erstmanifestation liegt zwischen dem 20.-40. Lebensjahr. Erkrankungen vor dem 15. Lebensjahr und nach dem 65.-70. Lebensjahr stellen eine Rarität dar.
→ IV: Z.T. besteht eine familiäre Häufung und eine HLA-DRB1 sowie HLA-DQB1-Assoziation.
→ Ätiologie: Bis heute ist genau Pathogenese nicht bekannt; man geht von einem multifaktoriellen Geschehen aus:
→ I: Genetische Faktoren: Eine familiäre Häufung ist bekannt; häufig besteht eine Assoziation zu HLA-DRB-1, HLA-DQB1 und/oder eine Punktmutation im BTNL2 kodierenden Gen.
→ II: Umweltfaktoren: Exposition mit spezifischen Trigger-Agenzien wie exogene Noxen wie Baumpollen, anorganische Substanzen (z.B. Aluminium), aber auch Viren und Mykobakterien werden diskutiert.
→ Pathogenese:
→ I: Charakteristischerweise entwickeln sich disseminierte, nicht-verkäsende, epitheloidzellige Granulome mit Langerhans-Riesenzellen. Diese weisen Einschlusskörperchen auf, zu denen die
→ 1) Schaumann-Körper und
→ 2) Asteroid-Körper gehören.
→ II: Ursache ist eine überschießende T-Helferzell-Typ-I-Reaktion mit konsekutiv vermehrter Produktion von Tumor-Nekrose-Faktor-Alpha (= TNF-Alpha), Interferon-y und Interleukin 2 durch Entzündungszellen, die die Granulomentstehung fördern. Zudem wandern Makrophagen ein und akkumulieren. Unter dem Einfluss der o.g. Mediatoren differenzieren sie sich zu Epitheloidzellen und fusionieren z.T zu Langerhans-Riesenzellen.
→ III: Pathologie: Bei der Sarkoidose manifestieren sich dichte kompakte Granulome sowie ein Nebeneinander von jungen Granulomen mit vielen mononukleären Zellen und alten zellarmen Granulomen (mit Hyalinisierung und Fibrose). Morphologisch charakteristisch für die Granulome der Sarkoidose sind 2 Zonen:
→ 1) Eine zentrale bzw. follikuläre Zone, die mit Makrophagen, mehrkernigen Riesenzellen und Epitheloidzellen dicht gepackt sind und
→ 2) Einer peripheren Zone, die einen lockeren Saum von Lymphozyten, Monozyten und Fibroblasten aufweist.
→ Klinik:
→ I: Allgemein: Die Sarkoidose ist eine Systemerkrankung und ihr klinisches Bild wird durch den jeweiligen Organbefall bestimmt. Allgemeinsymptome sind u.a.:
→ 1) Müdigkeit, Abgeschlagenheit, Fieber, Gewichtsverlust.
→ 2) Pulmonale Symptome: Beinhalten Husten, Dyspnoe, uncharakteristische Thoraxschmerzen (= Pleuradynie), selten Hämoptysen.
→ 3) Zumeist zeigt die Sarkoidose einen schleichenden Beginn.
→ II: Akute Sarkoidose: (= Morbus Löfgren) Selten in 10% der Fälle. Manifestiert sich vorwiegend bei junge Frauen um das 30. Lebensjahr mit akutem schwerem Krankheitsgefühl, Abgeschlagenheit, Schwächegefühl, Muskelschmerzen, Fieber, Husten und der klassischen Symptomatik, bestehend aus:
→ 1) Erythema nodosum mit initial roten im weiteren Krankheitsverlauf bräunlich-lividen druckschmerzhaften Knoten, bevorzugt an den Streckseiten der Beine,
→ 2) Arthritis (insbesondere im oberen Sprunggelenk) und
→ 3) Bihiläre Lymphknotenschwellung.
Laborchemisch lassen sich unspezifische Entzündungsparameter wie beschleunigte BSG bei (für den Morbus Löfgren charakteristisch) normalen ACE-Werten nachweisen. Innerhalb von 1-2 Jahren (zumeist im Zeitintervall von 6 Wochen) kommt es in fast 90% der Fälle zu einer Spontanremission, sodass in der Regel keine Therapie nötig ist.

→ III: Chronische Sarkoidose: (95%): Meist lang asymptomatisch, evtl. Müdigkeit, später Reizhusten und Dyspnoe (Zufallsbefund) und Fieber; die chronische Form kann aus einer einer akuten hervorgehen.
→ Klinisch-relevant: Klinisches Charakteristikum der Sarkoidose ist die große Diskrepanz zwischen dem schlechten radiologischen Befund und dem subjektiv guten Allgemeinzustand des Patienten.
→ IV: Sonderform EOS: (= Early-onset-sacoidosis) Es tritt ausschließlich vor dem 5. Lebensjahr mit einer klassischen Symptomtrias auf:
→ 1) Arthritis und Uveitis.
→ 2) Exanthem.
→ 3) Weitere Symptome sind Müdigkeit, Hepatomegalie, Fieber und Anorexie. Kann sporadisch oder familiär gehäuft auftreten.
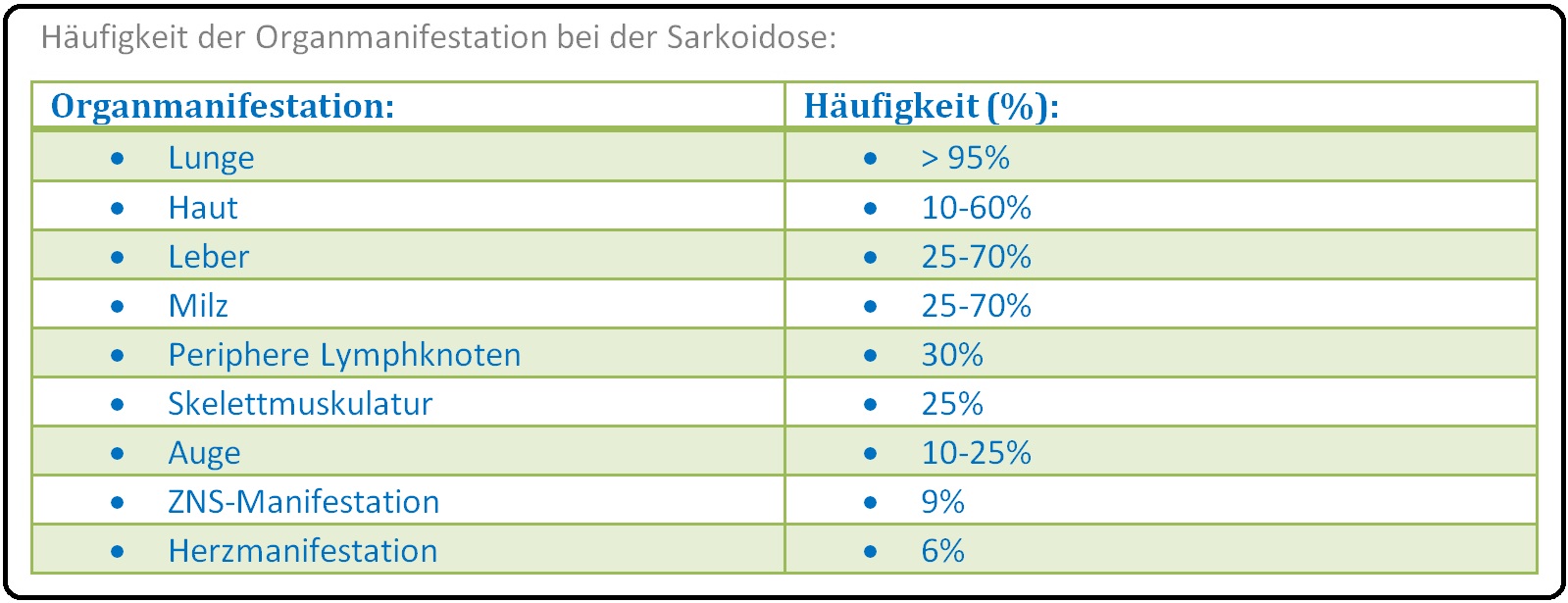
→ V: Extrapulmonale Manifestation:
→ 1) Haut:
→ A) Disseminierte klein- bis großknotige rotbraune Papeln.
→ B) Lupus pernio: Es handelt sich um livide Infiltrationen häufig im Bereich der Nase, Wangen und Ohrläppchen (durch Glasspateldruck werden die grauen Infiltrationsknötchen sichtbar).
→ C) Erythema nodosum: Subkutane rot-braune Knoten im Bereich der Streckseiten der unteren Extremität. Weitere (differenzialdiagnostische) Ursachen für das Erythema nodosum sind u.a. Infektionen (z.B. A-Streptokokken, Salmonellen, Shigellen, Yersinien), chronische Erkrankungen (z.B. Morbus Behcet, Colitis ulcerosa etc.), sowie Schwangerschaft im ersten Trimenon und nicht zuletzt idiopathisch.
→ 2) Auge: Mit Keratokonjunktivitis, Uveitis und Iridozyklitis.
→ 3) Knochen: Zystische Umwandlung der Fingerphalangen als Ostitis multiplex cystoides (= Jüngling-Syndrom), aber auch Störungen des Knochenmarks mit Panzytopenie und autoimmunhämolytischer Anämie.
→ 4) Heerfordt-Syndrom: Gekennzeichnet durch Parotitis, Uveitis und Faszialisparese.
→ 5) Kardiale Sarkoidose: Perikarderguss, Herzinsuffizienz, Myokarditis, Kardiomyopathie, Herzrhythmusstörung mit der Gefahr des plötzlichen Herztodes.

→ 6) Renale Folgeerkrankungen: Wichtige renale Folgeerkrankungen der Sarkoidose sind u.a. Kalziumstoffwechselstörungen, interstitielle Nephritis, Glomerulopathie (z.B. membranöse -, membranoproliferative -, fokal-segmental-sklerosierende Glomerulonephritis, etc.) selten und nicht zuletzt die retroperitoneale Sarkoidose mit obstruktiver Uropathie.
→ 7) ZNS-Sakroidose: Allgemein Hirnnervenparesen insbesondere die Faszialisparese, Polyneuropathie, zerebrale Endokrinopathie wie Hypophysenvorderlappeninsuffizienz, Diabetes insipidus, aber auch granulomatöse Meningitis oder Enzephalitis.
→ 8) Weitere Manifestationen: Hierzu zählen:
→ A) Hämatologische Störungen: Anämie und Lymphozytopenie.
→ B) Leberbefall: Granulomatöse Infiltrationen, Hepatomegalie.
→ C) Skelettmuskulatur: Insbesondere proximaler Muskelbefall im Bereich der Schulter und der Hüfte.
→ Komplikationen: Wichtige und z.T. schwerwiegende Komplikationen bei der Sarkoidose sind insbesondere:
→ I: Respiratorische Insuffizienz,
→ II: Cor pulmonale.
→ III: Plötzlicher Herztod bei Befall des kardialen Reizleitungssystems.
→ IV: Aber auch schwerwiegende neurologische Erkrankungen wie die Sinusvenenthrombose oder den Hirnabszess.
→ Diagnose:
→ I: Labor:
→ 1) In der akuten Phase (Morbus Löfgren), BSG-Erhöhung, Leukozytose.
→ 2) In 50% der Fälle manifestiert sich eine Gammaglobulinämie vom IgG-Typ.
→ 3) Hyperkalzämie und Hyperkalziurie durch vermehrte Produktion von 1,25-(OH2)-Vitamin-D3 in den Epitheloidzellen.
→ 4) Evtl. Leuko- und Lymphozytopenie
→ 5) Bestimmung der Aktivitätsparameter:
→ A) Angiotensin-Converting-Enzyme: (= ACE) Wird vor allem von den Epitheloidzellen der Granulome gebildet, sodass erhöhte ACE-Serumkonzentrationen als Maß der Granulomlast im Organismus gelten; und
→ B) Löslicher Interleukin-2-Rezeptors (sIL-2-R auch als Aktivitätsmarker). Dieser hat bei der Sarkoidose ein prognostischen Aussagewert, da bei der Hälfte der Betroffenen ohne akute Therapieindikation erhöhte Werte eine Behandlungsindikation innerhalb der nächsten 6 Wochen anzeigen.
→ II: Lungenfunktion: (= Lufu) Mit Nachweis einer restriktiven Ventilationsstörung und Einschränkung des CO-Transfers, die im weiteren Krankheitsverlauf eine obstruktive Kompotente entwickelt. Des Weiteren erfolgt die Blutgasanalyse in Ruhe und unter Belastung z.B. nach Treppensteigen (Normwerte der BGA), die den Grad der Funktionseinschränkung global erfasst, der 6-Minuten-Gehtest und nicht zuletzt die Spiroergometrie.
→ III: Bildgebende Verfahren:
→ 1) Röntgen Thorax: Und 67-Gallium-Szintigraphie (mit Anreicherung des Nukleotids in den pulmonalen und extrapulmonalen aktiven Granulomen z.B. der Lunge und Parotis, etc.)
→ 2) CT-Thorax: Insbesondere bei unklaren klinischen und/oder radiologischen Befunden. Bedeutende computertomographische Befunde sind u.a. mikronoduläre Verschattungen, die bevorzugt peribronchovaskulär lokalisiert sind, Konglomeratverschattungen, Verdickung der interlobulären Septen und nicht zuletzt eine gestörte Lungenarchitektur.
→ IV: Bronchoskopie: (Mit bronchoalveolärer Lavage) Nachweis einer lymphozytären Alveolitis mit erhöhtem CD4/CD8 Quotienten > 5 (Verschiebung des T-Helferzellen/T-Suppressorzellen-Quotienten zugunsten der T-Helferzellen; normal liegt er bei 2).
→ V: Transbronchiale Lungenbiopsie: Histologische Darstellung nicht-verkäsender Granulome (sehr sensitiv).
→ VI: Weitere Untersuchungen:
→ 1) Kardial: Ruhe-EKG, Langzeit-EKG, Belastungs-EKG und die Echokardiographie.
→ Klinisch-relevant: Aufgrund der potenziell lebensbedrohlichen lebensbedrohlichen Myokardsarkoidose, die nicht selten mit einem plötzlichen Herztod einhergeht, sollte bei unklarem AV-Block, weiteren Herzrhythmusstörungen oder einer dilatativen Kardiomyopathie immer auch an eine Sarkoidose gedacht werden.
→ 2) Neurologisch: MRT mit Liquoruntersuchung mit möglichem Nachweis einer Neurosarkoidose und konsekutiver Erhöhung von Lysozym, Mikroglobulin, ACE sowie einer lymphozytären Pleozytose.
→ 3) Augenuntersuchung.

→ Klassifikation: Der Sarkoidose nach dem Röntgen-Thorax-Befund:
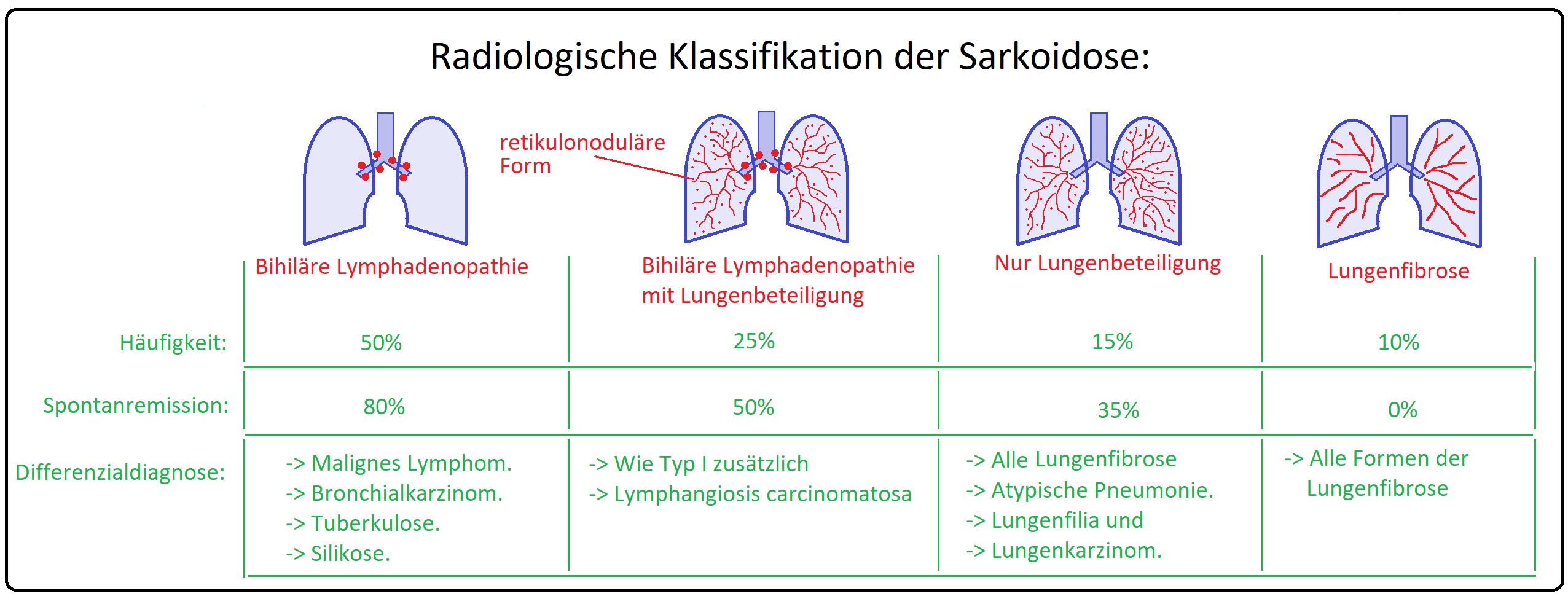
→ I: Typ 1: Bihiläre Lymphadenopathie mit vergrößerten polyzyklischen Lungenhili (Lunge ist nicht infiltriert, jedoch können andere Organe betroffen sein).
→ II: Typ 2: Bihiläre Lymphadenopathie mit klein- bis mittelfleckige pulmonale Infiltrationen (mit Lungengerüstveränderungen in den Lungenmittel- und Lungenoberfeldern.
→ III: Typ 3: Diffuse Lungeninfiltration ohne Lymphadenopathie.
→ IV: Typ 4: Irreversible Lungenfibrose mit retikulo-nodulärer Streifung (Honigwabenmuster der Lunge).

→ Differenzialdiagnose: Von der Sarkoidose müssen insbesondere nachfolgende Erkrankungen abgegrenzt werden:
→ I: Typ 1: Lungen-TBC, Bronchial-Ca, malignes Lymphom.
→ II: Typ 2/3: Allergische Alveolitis, Ornithose, Pneumokoniosen wie die Silikose oder Asbestose, Miliar-TBC, Alveolarzellkarzinom.
→ III: Typ 4: Lungenfibrose anderer Genese.
→ IV: Des Weiteren Morbus Behcet bzw. Neuro-Behcet. multiple Sklerose, aber auch Infektionen wie z.B. Lyme-Borreliose, etc.
→ Therapie: Als absolute Therapieindikationen gelten schwere Allgemeinsymptome, Hyperkalzämie, Hyperkalziurie, deutliche Störungen der Lungenfunktion, Lupus pernio, Ikterus, Myokard-, Nieren- und ZNS-Beteiligung.
→ I: Akute Form: Symptomatische Behandlung mit NSAR bzw. Analgetika wie z.B. Diclofenac.
→ II: Chronische Form:
→ 1) Eine Kortikosteroid-Therapie ist indiziert:
→ A) Ab Typ 2 mit Verschlechterung der Lungenfunktion,
→ B) Hyperkalzämie oder Hyperkalziurie,
→ C) Beteiligung von Augen, Myokard, Niere oder ZNS.
→ D) Dosierung: 20-50mg/d Prednisolon über 1-3 Monate (unter radiologischer Kontrolle), danach stufenweise Reduktion um 5-10mg/Monat auf eine Erhaltungsdosis von 7,5-15mg/d. Ein Auslassversuch kann nach 6-12 Monaten versucht werden.
→ 2) Kombinationstherapie: Bei unzureichender Wirkung von Prednisolon bzw. Progression der Erkrankung kann das Kortikosteroid mit Methotrexat oder Anti-TNF-Alpha kombiniert werden.

→ Prognose:
→ I: Die akute Sarkoidose mit fieberhaften Krankheitsbild hat eine gute Prognose.
→ II: Bei der chronischen Verlaufsform mit multiplen Organmanifestationen weisen nur 1/3 der Patienten ein Restitutio ad integrum auf, bei einem weiteren 1/3 kommt es zu einer Befundverbesserung und in 1/3 der Fälle lässt sich ein chronisch-progredienter Krankheitsverlauf dokumentieren.
→ III: Die Letalität der Sarkoidose liegt < 5%.