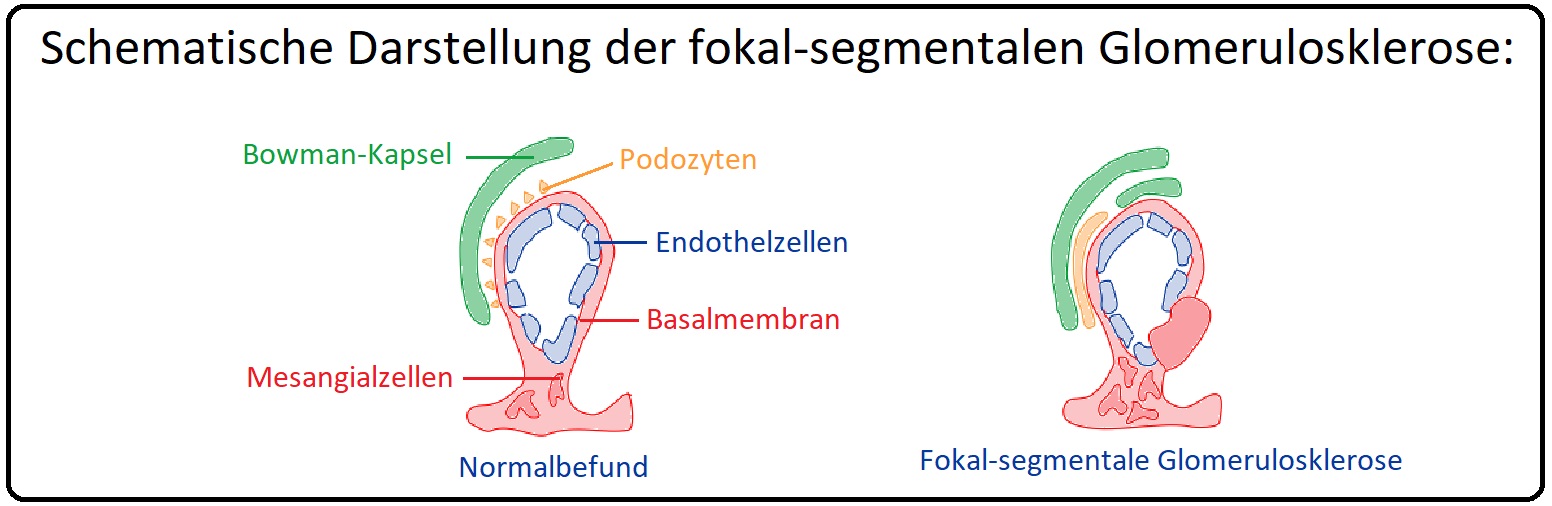
→ Definition: Bei der (primär) fokal-segmentalen Glomerulosklerose handelt es sich um ein klinisch-histologisches Syndrom, gekennzeichnet durch eine fokale segmentale Sklerose der Glomeruli, Verschmelzung der Fußfortsätze der Podozysten und der damit korrelierenden typischen Symptomatik des nephrotischen Syndroms.
→ Epidemiologie:
→ I: Im Erwachsenenalter stellt die fokal-segmental-sklerosierende Glomerulonephritis die dritthäufigste Ursache für das nephrotische Syndrom dar.
→ II: Es zeigt sich sowohl eine familiäre Häufung als auch eine erhöhte Rezidivrate nach Nierentransplantation.
→ Ätiopathogenese:
→ I: Primäre FSGN: Pathogenetisch kommt es bei der primären Form zu einer Läsion der glomerulären Podozyten (= Epithelzellen). Die Zellen sitzen mit ihren Fortsätzen auf der glomerulären Basalmembran.
→ 1) Wie bei der Minimal-change-Glomerulonephritis wird bei der idiopathischen Form auch eine zytokinvermittelte immunologische Ursache vermutet. Für einen solchen zirkulierenden die Podozyten schädigende Permeabilitätsfaktor spricht, dass es nach Nierentransplantation in bis zu 30% der Fälle zu einem Rezidiv kommen kann.
→ 2: Als Permeabilitätsfaktoren werden u.a. der Soluble Urokinase Plasminogen Rezeptor (suPAR bei Kindern in 55% der Fälle und bei Erwachsenen bei 75% der Fälle liegt), das Cardiotrophin-like-Zytokin1 (CLZ-1 aus der Familie des Interleukin 6) und nicht zuletzt TNF-Alpha diskutiert.
→ II: Bei den selten familiären und kongenitalen Formen der fokal-segmental-sklerosierenden Glomerulonephritiden zeigen sich Mutationen an Genen für z.B. Podozytenprotein wie Nephrin, Alpha-Aktin-4, Podozin, CD-2-Adapterprotein, etc.
→ III: Sekundäre FSGN: Bei dieser Form wird eine Dysbalance zwischen Anforderungen und Leistungsfähigkeit der Glomeruli angenommen (= Hyperfiltration).
→ IV: Eine Sonderform der fokal-segmentalen Glomerulonephritis ist die APOL-1-assoziierte Form, die überwiegend bei Patienten afrikanischen Ursprungs auftritt und durch eine genetische Variation des APOL-1-Gens charakterisiert ist.
→ Klassifikation: Die fokal-segmental-sklerosierende Glomerulonephritis wird unterteilt in:

→ Klinik:
→ I; Klinisch imponierte die fokal-segmentale Glomerulosklerose häufig durch das Vollbild des nephrotischen Syndroms (70-80% der Fälle) mit ausgeprägten generalisierten Ödemen, nicht-selektiver Proteinurie, Hypalbuminämie und Hypoproteinämie, etc.
→ II: Nicht selten manifestiert sich auch eine Hämaturie sowie (renale) arterielle Hypertonie.
→ III: Aber auch ein asymptomatischer Krankheitsverlauf mit alleiniger Proteinurie ist möglich.
→ Diagnose:
→ I: In der Anamnese lässt sich häufiger eine vorausgehende Erkrankung der oberen Atemwege eruieren (die Korrelation ist noch nicht bekannt). Auch zeigt sich nicht selten während der Schwangerschaft ein erstmaliges Auftreten der fokal-segmentalen Glomerulosklerose bzw. eine Progredienz der Erkrankung. Bei der klinischen Untersuchung kann evtl. eine renale Hypertonie nachgewiesen werden.
→ II: Labor: Nachweis einer Proteinurie (> 3g/24h im nephrotischen Bereich), evtl. Hämaturie, die z.T. makroskopisch imponieren kann, sowie Nierenfunktionsstörung mit konsekutiver Kreatininerhöhung (in 20-25% zur Diagnosestellung).
→ III: Pathohistologie: Die Diagnose-Sicherung der fokal-segmental sklerosierenden Glomerulonephritis erfolgt histologisch:
→ 1) Mikroskopisch sind fokal und segmental-sklerosierende glomeruläre Veränderungen mit kollabierten Kapillaren und Adhäsionen zwischen den Kapillarschlingen und der Bowman-Kapsel.
→ 2) Elektronenmikroskopisch ist eine Fusion aller Podozytenfortsätze an den Glomeruli darstellbar (wie bei der Minimal-change-GN).
→ 3) Die fokal-segmental-sklerosierende Glomerulonephritis lässt sich histologisch in weitere 5 Subtypen unterteilen:

→ Differenzialdiagnose: Von der fokal-segmental-sklerosierenden Glomerulonephritis müssen insbesondere nachfolgende Erkrankungen abgegrenzt werden:
→ I: Klinische und histologische Unterscheidung zwischen den Formen der fokal-segmental-sklerosierenden Glomerulonephritis (primären -, sekundäre sowie familiäre kongenitaler FSGS).
→ II: Minimal-Change-Glomerulonephritis und
→ III: Weitere Erkrankungen, die mit einem nephrotischen Syndrom einhergehen z.B. membranöse Glomerulonephritis, membranoproliferative GN, diabetische Nephropathie, C3-Glomerulopathie, Plasmozytom, Nierenvenenthrombose, systemische Amyloidose, etc.
→ Therapie: Die Therapie der fokal-segmentalen Glomerulosklerose ist durch die geringe spontane Remission (< 5% der Fälle) und des häufig progredienten Nierenfunktionsverlustes z.T. sehr problematisch.
→ I: Allgemein:
→ 1) Bei Patienten mit guter Prognose (Proteinurie < 3,5g/d) reicht zumeist eine alleinige symptomatische Behandlung mit ACE-Hemmern oder AT1-Rezeptor-Antagonisten.
→ 2) Bei der primär idiopathischen Form steht die immunsuppressive Therapie mit Glukokortikoiden (1mg/kgKG/d) im Vordergrund.
→ 3) Insbesondere bei der sekundären Form ist die Behandlung der Grunderkrankung von besonderer Bedeutung.
→ II: Steroidmonotherapie: Sie wird v.a. bei Patienten mit primärer FSGS mit nephrotischem Syndrom.
→ 1) Die Dosis von 1mg/kgKG/d (maximal 80mg/d) wird über mehrere Monate (maximal 4 Monate) bis zum Erreichen einer vollständigen Remission appliziert. Anschließend erfolgt eine schrittweise Reduktion der Glukokortikoid-Dosis über einen Zeitraum von 6 Monaten (zunächst 10mg alle 2 Wochen, danach alle 2-4 Wochen um 2,5mg).

→ 2) Bei häufigen Rezidiven wird eine Kombinationstherapie aus Cyclosporin A (3mg/kgKG/d) oder Tacrolismus (0,05-0,1mg/kgKG/d) mit Prednisolon (0,15mg/kg KG/d) über 4-6 Monate empfohlen; die anschließende Dosisreduktion über 4-8 Wochen.
→ III: Steroidresistenz: Eine Steriodresistenz bei der FSGS ist definiert als eine Persistenz des nephrotischen Syndroms trotz hochdosierter Glukokorikoidtherapie über 6 Monate. Hierbei sind die Behandlungsoptionen eingeschränkt. Cyclophosphamid ist kaum wirksam und Cyclophosphamid weist auch keine optimale Wirkung auf.
→ 1) Bei 60% der mit Cyclosporin behandelten Patienten kommt es zur Remission (bei 20% erfolgt eine vollständige, bei 40% eine partielle Remission).
→ 2) Jedoch zeigt sich nach Absetzen des Cyclosporins in 60% der Fälle ein Rezidiv, sodass die Therapie mit Cyclosporin A nach Erreichen einer (partiellen bzw. kompletten) Remission für mindestens 12 Monate fortgesetzt werden sollte (anschließende Reduktion um 25% alle 2 Monate).
→ 3) Sollte jedoch keine Remission mit Cyclosporin A nach 6 Monaten erreicht werden, wird eine Beendigung dieser Behandlung empfohlen.
→ IV: Nierentransplantation: Bei Entwicklung einer terminalen Niereninsuffizienz im Rahme einer fokal segmental-sklerosierenden Glomerulonephritis ist die Nierentransplantation indiziert. Hierbei zeigt sich in bis zu 30% der Fälle ein Rezidiv im Transplantat, sodass eine Plasmapherese und Immunabsorption zur Entfernung der Permeabilitätsfaktoren erwogen werden sollte (Remissionen sind möglich, häufiger aber nur von begrenzter Dauer).
→ Prognose: Die Langzeitprognose der fokal-segmental sklerosierenden Glomerulonephritis ist von 2 Parametern abhängig nämlich vom Ausmaß der Proteinurie und dem Ansprechen der Proteinurie auf Glukokortikoide.
→ I: Günstige Langzeitprognose: Günstige renale Prädiktoren sind:
→ 1) Nachweis einer Proteinurie < 3,5g/d (20% weisen eine terminale Niereninsuffizienz nach 10 Jahren auf).
→ 2) Nachweis einer kompletten (90%iges renales 10-Jahresüberleben) oder partiellen Remission (78% iges renales 10-Jahresüberleben). Bei keiner Remission liegt das renale 10 Jahresüberleben bei nur noch 40%.
→ II: Ungünstige Langzeitprognose: Ungünstige Prädilektoren mit progredientem Nierenfunktionsverlust sind insbesondere:
