→ Definition: Beim Lambert-Eaton-Syndrom handelt es sich um eine paraneoplastische oder autoimmunologische Störung der präsynaptischen Acetylcholin-Ausschlüttung durch Antikörper-vermittelte Blockade der spannungsabhängigen Kalzium-Kanäle (P/Q-Typ) mit konsekutiver Beeinträchtigung der Signalübertragung an der neuromuskulären Endplatte.
→ Epidemiologie:
→ I: Die Prävalenz für die Entwicklung eines Lambert-Eaton-Syndroms liegt in Deutschland bei 1/100000 Einwohnern, wobei insbesondere Männer (M : F = 5: 1; im Gegensatz zur Myasthenia gravis) betroffen sind.
→ II: Es kann in jedem Alter auftreten, jedoch besteht ein Manifestationsgipfel im mittleren bis höheren Lebensalter, zumeist nach dem 60. Lebensjahr.
→ Ätiopathogenese:
→ I: Charakteristikum bei der Genese ist eine Kreuzreaktion von IgG-Antikörpern gegen spannungsabhängige präsynaptische Kalziumkanäle peripherer Nerven (= voltage-gated-calcium-channels = VGCC vom Typ P/Q in > 80% der Fälle). Folge ist eine deutlich verringerte Freisetzung von Acetylcholin mit konsekutiver Beeinträchtigung der motorischen Endplatte.
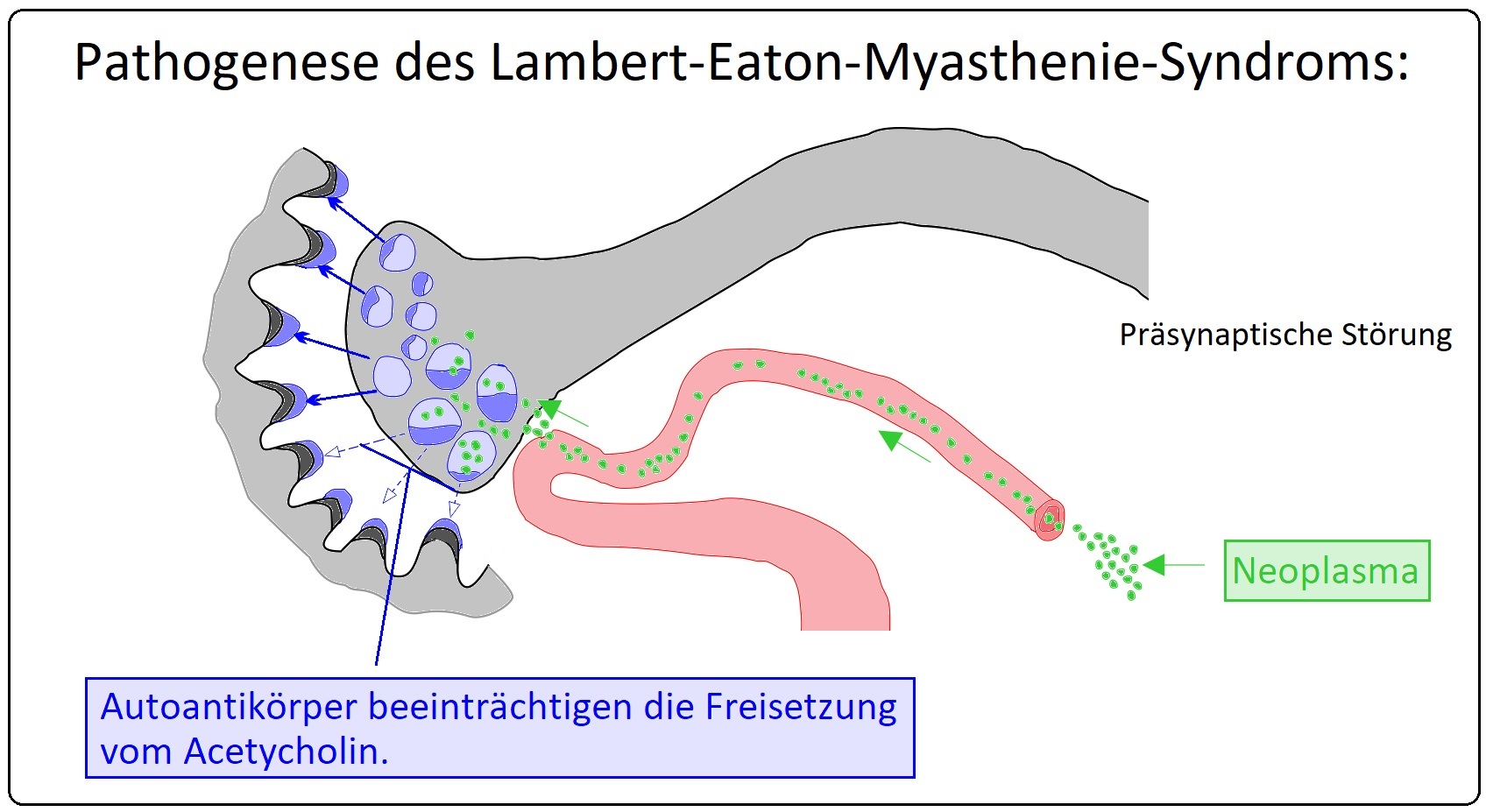
→ II: Ursachen: Sind insbesondere:
→ 1) Paraneoplastisch: Bei Malignomen v.a. das kleinzellige Bronchialkarzinom (in 2/3 der Fälle) seltener beim NSCLC, Prostata-Ca, Lymphomen, Neuroblastom oder Andenokarzinomen der Mamma, des Magens, etc. Hierbei ist oftmals ein zusätzlicher Nachweis von Hu-, CV2/CRMP- und/oder SOX-1-Antikörpern möglich.
→ 2) Autoimmunologisch: Vermehrte Auftreten bei einigen weiteren Erkrankungen wie die Autoimmun-Thyreoiditis (z.B. Hashimoto-Thyreoiditis, Morbus Basedow), perniziöse Anämie, rheumatoider Arthritis, Sjögren-Syndrom, Vitiligo, Zöliakie, Colitis ulcerosa, etc. und einer signifikanten Assoziation mit HLA-B8 sowie -DR3.
→ Klinik:
→ I: Im Mittelpunkt dieser myasthenie-ähnlichen Erkrankung stehen Schwäche und vorzeitige Ermüdbarkeit der proximalen Extremitätenmuskulatur, insbesondere des Beckengürtels und der Oberschenkel, sowie Abschwächung bzw. Verlust der Muskeleigenreflexe. Typisch hierbei ist die initial vorübergehende Zunahme der Muskelkraft, die sich dann anschließend progredient verringert. Eine Muskelatrophie fehlt in der Regel.
→ II: Hirnnervenausfälle: Mit Ptosis, Augenmuskelparesen, Doppelbildern, Akkommodationsstörungen, aber auch z.B. Dysphagie sowie Sprechstörungen etc. (häufig erst im späteren Krankheitsverlauf oder gar nicht).
→ III: Des Weiteren evtl. Auftreten von Myalgien und Parästhesien sowie einer begleitenden paraneoplastischen Kleinhirndegeneration (selten).
→ IV: Autonome Symptome: Bei etwa der Hälfte der Patienten zeigen zusätzlich cholinerge Symptome mit:
→ 1) Blasen- und Potenzstörungen,
→ 2) Mangelnde Speichel- und Schweißsekretion und nicht zuletzt
→ 3) Orthostatische Hypotonie (insbesondere bei Nachweis von Anti-Hu-Antikörpern) sowie
→ 4) Gewichtsverlust und Obstipation.
→ Diagnose:
→ I: Anamnese: Exploration des Beschwerdebeginns und Tagesverlaufs sowie der Symptomatik unter Belastung.
→ II: Klinische Untersuchung:
→ 1) Klinisch-neurologische Test wie das wiederholte Öffnen und Schließen der Augenlider, Hände, das Anheben des Kopfes im Liegen etc. mit konsekutivem Nachweis einer raschen Ermüdbarkeit.
→ 2) Reflexbahnung nach Willkürkontraktion; typisch ist ein fehlender Eigenreflex in Ruhe und anschließender Auslösung oder Steigerung nach 20-30 sec. maximaler Willkürinnervation.
→ 3) Tensilon-Test: (= Nach intravenöser Injektion von 5-10mg von Edrophoniumchlorid, ein Cholesterinesterase-Hemmer, Besserung der klinischen Symptomatik nach 30-60sec. Nach Applikation und nach 5min wieder rückläufig). Meist negativ oder nur leicht positiv.
→ III: EMG:
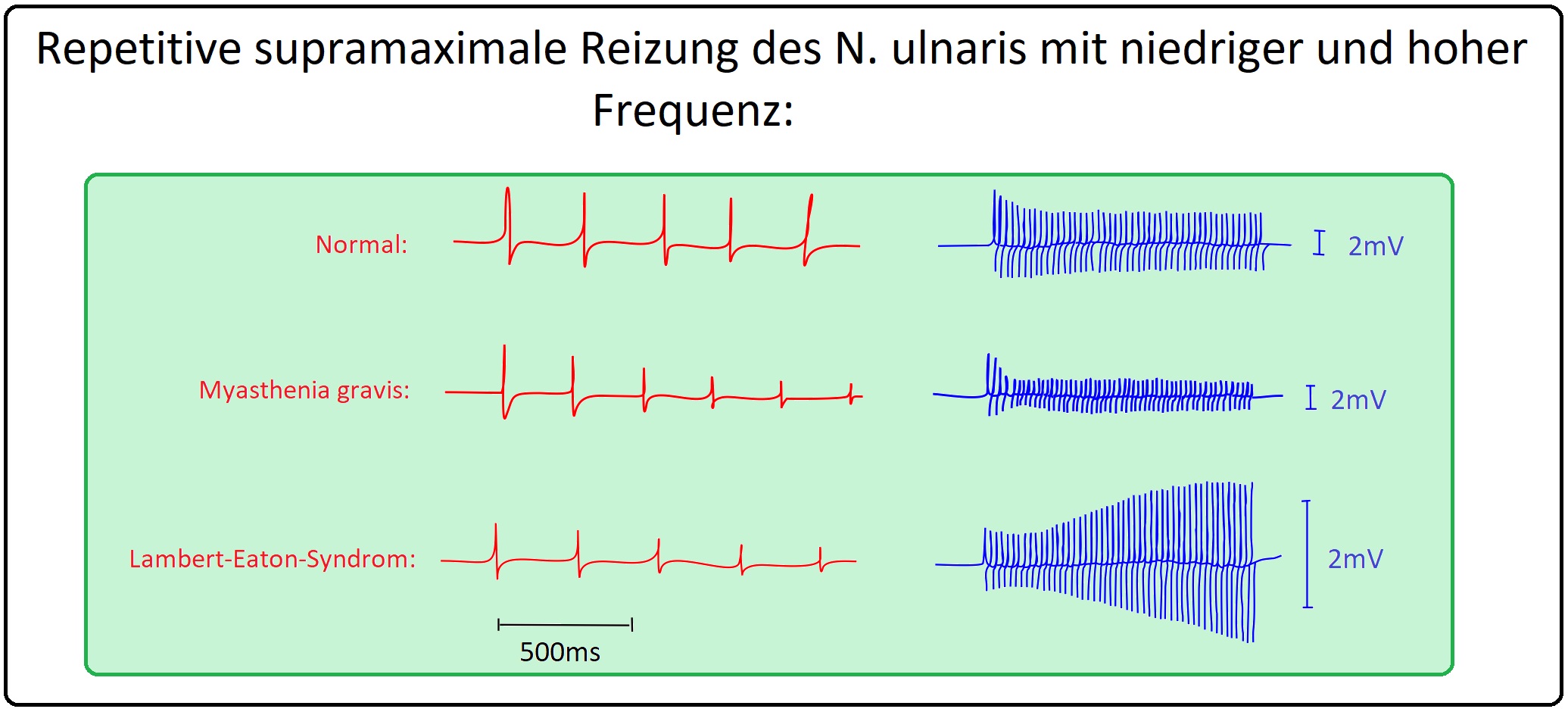
→ 1) Hochgradige Amplitudenminderung des Muskelaktionspotenzials in Ruhe; Zunahme um > 200% nach 20-30sec. maximaler Willkürinnervation.
→ 2) Ermüdungstest: Die repetitive Stimulation mit 20-30 Hz. führt im Unterschied zur Myasthenia gravis vorübergehend zur Amplitudenzunahme (wird als Inkrement bezeichnet). Bei wiederum niedrigen Stimulationsfrequenzen (3-5 Hz) nehmen die schon ohnehin niedrigen Muskelantwortpotenziale weiter ab (= Dekrement).
→ IV: Labor: Möglicher Nachweis der verschiedenen Antikörper gegen spannungsgesteuerte Kalziumkanäle (= VGCC), SOX-1, AGNA (= Anti-Glial-Nuclear-Antibody), die als Marker insbesondere für das SCLC-LEM-Syndrom gelten.
→ V: Bildgebung: Insbesondere CT und FDG-PET dienen der Tumorsuche. Bei initial negativem Befund wiederholte halbjährliche radiologische Kontrollen über ein Zeitintervall von mindestens 2 Jahren.
→ Differenzialdiagnose: Vom Lambert-Eaton-Myasthenie-Syndrom müssen insbesondere nachfolgende Erkrankungen abgegrenzt werden:
→ I: Myasthenia gravis,
→ II: Guillain-Barre-Syndrom,
→ III: Familiäres Hypokaliämie-Syndrom mit periodischer Lähmung,
→ IV: Thyreotoxikose und nicht zuletzt
→ V: Die cholinerge Krise.
→ Therapie:
→ I: Ursächliche Therapie: Bei der paraneoplastischen Form führt die Tumorentfernung häufig zu einer deutlichen Rückbildung der klinische Symptomatik. Beim autoimmunologischen Lambert-Eaton-Myasthenie-Syndrom sollte eine Behandlung mit Glukokortikoiden (z.B. Prednisolon 1,5mg/kgKG) oder Azathioprin 2,5mg/kgKG) versucht werden.
→ II: Symptomatische Therapie: Bei dieser Behandlung haben sich insbesondere 2 Präparate etabliert:
→ 1) 3,4-Diaminopyridin (Amifampridin) stellt einen reversiblen K-Kanal-Blocker dar und wird initial in einer Dosierung von 15mg/d mit langsamer Steigerung von 5mg alle 4-5 Tage auf eine Maximaldosis von 60-(100)mg/d verabreicht.
→ 2) Bei ausgeprägter klinischer Symptomatik können Immunglobuline i.v. bzw. eine Plasmapherese durchgeführt werden.
→ Prognose: Die Prognose des LEMS ist insbesondere von der Grunderkrankung abhängig und bei der paraneoplastischen Form zumeist sehr schlecht.