→ Definition: Beim Guillain-Barre-Syndrom handelt es sich um eine akut oder subakut verlaufende Polyradikuloneuritis, die innerhalb weniger Tage (bis Wochen) zu einer rasch aufsteigenden motorischen Parese (Tetraparese mit möglicher Ateminsuffizienz) mit geringen Sensibilitätsstörungen führen kann. Typischerweise beobachtet man eine multifollikuläre Demyelinisierung im Bereich peripherer Nerven und deren Wurzeln.
→ Epidemiologie:
→ I: Die Inzidenz des GBS liegt zwischen 0,5-2/100000 Einwohnern.
→ II: Die Krankheit kann in jedem Lebensalter auftreten, weist jedoch 2 Manifestationsgipfel zum einen zwischen dem 20.-30. und dem 50.-60. Lebensjahr auf, wobei das männliche Geschlecht überwiegt.
→ Ätiopathogenese:
→ I: Die Ursache ist bis heute noch nicht genau bekannt; man vermutet jedoch eine Autoimmunreaktion sowohl gegen periphere Nerven (= Polyneuritis) als auch deren Vorder- und Hinterwurzeln (Polyradikulitis) nach Infektionserkrankungen (seltener nach Impfungen). Wichtige Krankheitserreger hierbei sind u.a.: Varizella-Zoster-, Mumps-, Epstein-Barr-Virus, Zytomegalie-Virus, HIV, sowie Mykoplasmen und Campylobacter jejuni (selten aber auch infolge massiver Stresssituationen wie große Operation und Schwangerschaft).

→ II: Pathogenetisch wird eine durch Infektion getriggerte Immunreaktion (= zellvermittelter Typ mit humoraler Veränderung) angenommen. Sie bewirkt eine Kreuzreaktion mit konsekutiver Antikörperbildung gegen peripheres Myelin und die Axonmembran im Sinne einer melocular-mimicry (= fehlgeleitete Immunreaktion).
→ Klassifikation: Beim Guillain-Barre-Syndrom unterscheidet man verschiedene Subtypen mit typischen klinischen, elektrophysiologischen und immunologischen Charakteristika:
→ I:

→ II: Des Weiteren existierten noch weitere sehr seltene Subtypen wie die akute Pandysautonomie (mit fast ausschließlicher Beteiligung des sympathischen und parasympathischen Nervensystems sowie gehäufter Assoziation mit dem Epstein-Barr-Virus), das rein ataktische Guillain-Barre-Syndrom oder die isolierte Bulbärlähmung.
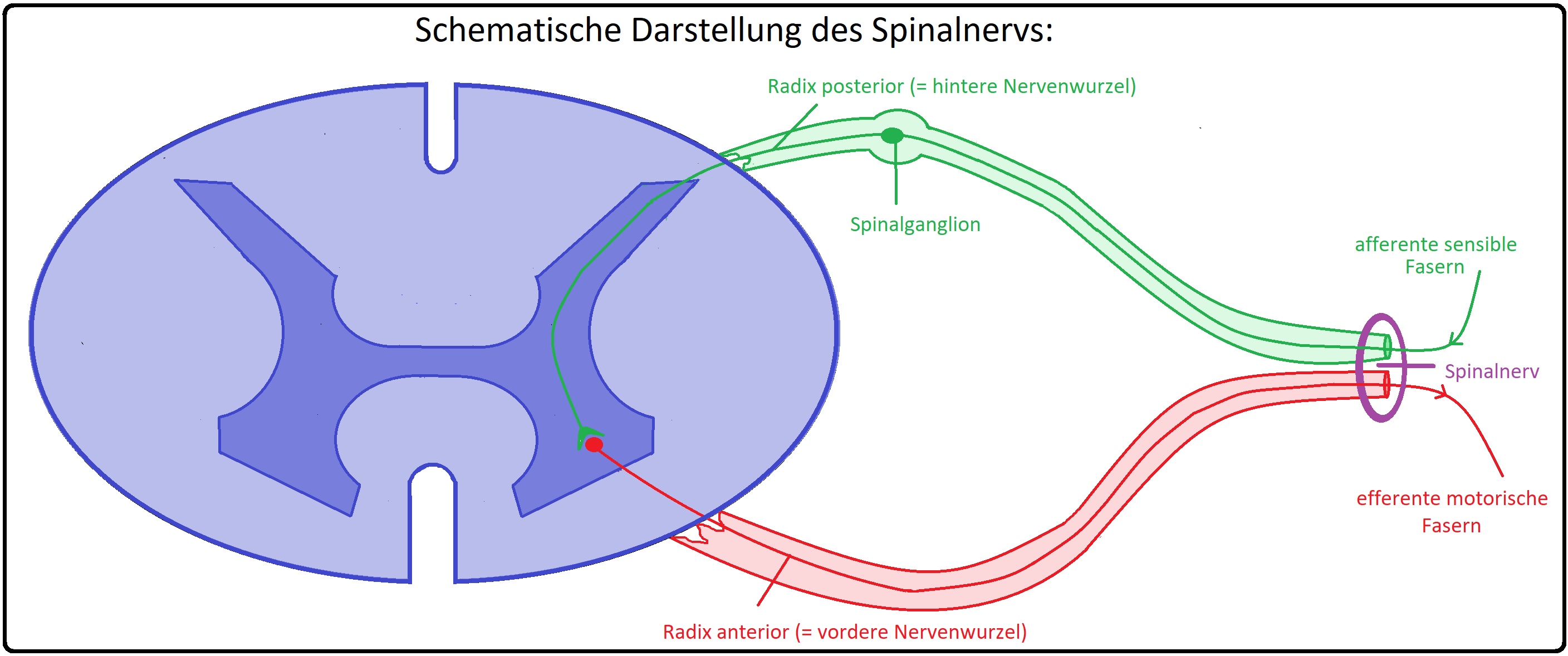
→ Pathologie: Es zeigen sich Infiltration und entzündliche Läsionen mit Rundzellen und Makrophagen in den Markscheiden mit konsekutiver Markscheidendegenerationen (Myelinzerfall). Betroffen sind insbesondere die vordere und hintere Nervenwurzel des Rückenmarks (= Polyradikulitis) sowie der dazugehörige periphere Nerv (= Polyneuritis).
→ Klinik: Zumeist kommt es 2-4 Wochen nach einem vorangegangenen Infekt der oberen Atemwege (bzw. des Gastrointestinal-Traktes) zu heftigen Rücken- und Gliederschmerzen sowie zu akrodistalen Parästhesien und Taubheitsgefühl insbesondere in den unteren Extremitäten.
→ I: Paresen: Symmetrische Schwäche der Beine, die sich von distal nach proximal ausbreitet und evtl. auf die Muskulatur des Rumpfes übergreifen kann. Die Ausprägung der motorischen Parese ist sehr variabel und reicht von nur leichten Paresen der Fußheber, Knie- und Hüftbeuger bis hin zur Tetraplegie. Bei Befall des Diaphragmas kann es zu Atemstörungen kommen.
→ II: Hirnnervenlähmung: (= Neuritis cranialis) Häufig manifestiert sich eine Hirnnervenbeteiligung mit z.B. Kau- und Schluckstörungen und Diplegia facialis (im Extremfall Locked-in-Syndrom); es betrifft insbesondere den Nervus facialis (in 50% der Fälle manifestiert sich Fazialisparese häufig beidseitig), N. trigeminus, N. vagus, N. accessorius und N. hypoglossus.
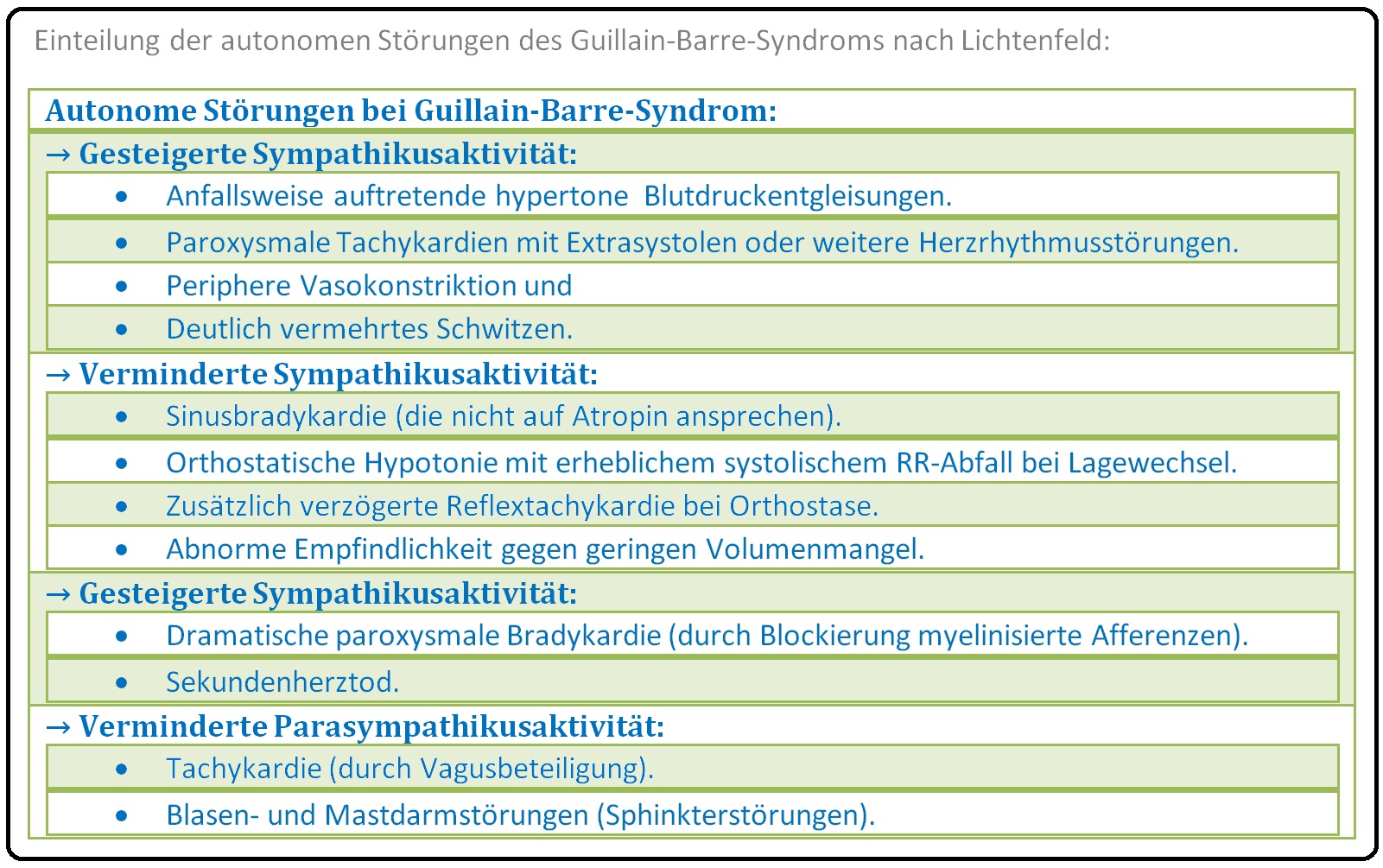
→ III: Autonome Störungen: Durch Befall afferenter und efferenter autonomer Fasern (Einteilung nach Lichtenfeld):
→ IV: Weitere Symptome: Sind u.a.:
→ 1) Verlust der Muskeleigenreflexe,
→ 2) Sensible Ausfälle zumeist nur von geringem Grade; sie können auch auf die Rumpfmuskulatur übergreifen.
→ 3) Gesteigerte oder aber auch verminderte ADH-Sekretion mit Entwicklung einer SIADH bzw. eines Diabetes insipidus.
→ Krankheitsverlauf:
→ I: Die Krankheit bildet sich zumeist akut oder subakut über Tage bis Wochen aus. Die Rückbildung der Klinik beginnt zumeist 2-4 Wochen nach Stillstand der Symptomatik und führt nicht selten zu einer Restitutio ad integrum. Es existieren aber auch chronisch protrahierte Krankheitsverläufe über Monate (bis Jahre).
→ II: Eine weitere Verlaufsform ist die perakute Entwicklung der Symptomatik, bei der der Patient am Morgen noch unauffällig ist und nachts in der Klinik schon intubiert und beatmet werden muss. Dies wird als Landry-Paralyse bezeichnet.

→ Komplikationen: Wichtige und z.T. schwerwiegende Komplikationen beim Guillain-Barre-Syndrom sind u.a.:
→ I: Infolge einer autonomen Neuropathie können sich Herzrhythmusstörungen sowie Asystolie und Herzversagen durch Überleitungsstörungen entwickeln.
→ II: Durch Immobilisation tiefe Beinvenenthrombose und Lungenembolie, aber auch Inaktivitäts- oder Pneumonie durch Aspiration.
→ III: Atemlähmung mit konsekutiver Beatmungspflicht und ggf. späteren Schwierigkeiten bei der Entwöhnung vom Respirator.
→ Diagnose:
→ I: Anamnese/klinische Untersuchung: Exploration vorangegangener Infekte (unspezifische Atemwegsinfekte oder Infekte des GIT insbesondere mit Campylobacter jejuni) sowie Nachweis der charakteristischen klinischen Symptome mit progredient symmetrisch aufsteigenden schlaffen Paresen, Reflexabschwächung bis hin zur Areflexie bei zumeist diskreten Sensibilitätsstörungen (Tiefensensibilität durch Lagesinn- oder Stimmgabeltest).
→ II: Labor:
→ 1) Erhöhung von CRP und Leukozytose sowie vermehrtes Auftreten von IgM, IgG und IgA in der Immunelektrophorese.
→ 2) Auf die Bestimmung der Antikörper wird zumeist verzichtet, kann aber im Einzelfall (v.a. bei schwieriger Differenzialdiagnose) wichtig sein.

→ 3) Liquor: Bei der Untersuchung zeigt sich ein klarer Liquor mit normaler Zellzahl (evtl. leichte Pleozytose 10 Zellen/mm3) bei deutlich erhöhtem Eiweiß (bis > 1000mg/dl infolge einer Blut-Hirn-Schrankenstörung); dies wird zytoalbuminäre Dissoziation bezeichnet (tritt häufig erst in der 2.-4. Krankheitswoche auf).
→ Klinisch-relevant: Eine Pleozytose mit > 50 Zellen und der Nachweis von Granulozyten im zytologischen Befund lassen an der Diagnose Guillain-Barre deutlich zweifeln.
→ III: Elektroneurographie:
→ 1) NLG: Hinweis für eine Demyelinisierung ist eine der motorischen Nervenleitungsgeschwindigkeit (= NLG) Verlangsamung bis hin zum Leitungsblock sowie eine verlängerte F-Wellen-Latenz.
→ 2) EMG: Elektromyographischer Nachweis von pathologischen Spontanaktivitäten als Ausdruck axonaler Degeneration zumeist 2-3 Wochen nach Erkrankungsbeginn.
→ IV: Evtl. Nervenbiopsie des N. suralis mit Demyelinisierung peripherer Nerven.
→ V: Vegetative Diagnostik: Hierbei erfolgen Untersuchungen zur Herzfrequenzvariabilität bei tiefer In- und Exspiration sowie Herzfrequenz und Blutdruckvariabilität bei Lagewechsel, aber auch Karotissinusmassage und der Bulbusdruckversuch (unter intensivmedizinischer Beobachtung).

→ Differenzialdiagnose: Vom Guillain-Barre-Syndrom müssen insbesondere nachfolgende Erkrankungen abgegrenzt werden:
→ I: Lubago-Ischias-Syndrom: Der Bandscheibenprolaps weist eine ähnliche Symptomatik mit Rückenschmerzen und radikulären Parästhesien auf.
→ II: Bei der Lyme-Borelliose nach Zeckenbiss tritt häufig eine klinische Symptomatik mit Polyradikulitis und sensomotorischen Ausfällen auf.
→ III: Autoimmunvaskulitis wie das Churg-Strauss-Syndrom oder die Panarteriitis nodosa (Abdominalsymptome).
→ IV: Toxische Polyneuropathie beim Critical-illness-Polyneuropathy (nach Sepsis), Polyradikulitis bei AIDS, aber auch bei der akuten intermittierenden Porphyrie, etc.
→ V: Sarkoidose: Mit typischer Zellzahlerhöhung im Liquor sowie charakteristischen radiologischen Veränderungen.
→ VI: Lambert-Eaton-Myasthenie-Syndrom sowie die Myasthenia gravis und nicht zuletzt
→ VII: Poliomyelitis acuta: Hierbei besteht immer ein asymmetrischer Befall und Sensibilitätsstörungen fehlen und nicht zuletzt dem
→ Therapie: Aufgrund der Gefahr der Atemlähmung und akuter Störungen des Herz-Kreislaufes werden Patienten mit Guillain-Barre-Syndrom intensivmedizinisch überwacht.
→ I: Symptomatische Therapie:
→ 1) Kontraktur-, Pneumonie und nicht zuletzt die Thromboseprophylaxe,
→ 2) Beatmung bei zunehmender Ateminsuffizienz mit Abfall der Vitalkapazität auf 25% des Normalwertes oder schnell aszendierenden Formen.
→ Klinisch-relevant: Die 3x tägliche Messung der respiratorischen Vitalkapazität bei Patienten mit Guillain-Barre-Syndrom ist obligat, um eine Dekompensation mit Beatmungspflichtigkeit nicht zu übersehen.

→ 3) Gegebenenfalls Schmerztherapie mit Antiphlogistika oder Opiaten.
→ 4) Autonome Störungen: Bei arterieller Hypertonie Gabe z.B. von Nifedipin oder eines Alpha-2-Rezeptoragonisten wie Clonidin, etc. bei Tachykardie Propranolol und bei Sinusbradykardie oder AV-Block II und III. Grades (EKG-Befund: AV-Block) Anlage eines passageren Schrittmachers.
→ II: Medikamentöse Therapie: Immunglobuline und Plasmapherese sind gleich wirksam.
→ 1) Immunglobuline: 7S-Immunglobuline sind wenig belastend und ohne wesentliche Nebenwirkungen. Die übliche Dosierung liegt bei 0,4g/kgKG pro Tag über mindestens 5 Tage. Vor allem muss auf ausreichende Flüssigkeitszufuhr geachtet werden (eine Kontraindikation ist der IgA-Mangel mit vermehrtem Risiko der Anaphylaxie).
→ 2) Plasmapherese: Sie ist insbesondere bei Kontraindikation für die Immungobulintherapie sowie bei rasch fortschreitenden Krankheitsverläufen indiziert bzw. gut wirksam. Hierbei wird jeden 2. Tag 40-50ml Serum/kgKG gegen Plasmaersatz (z.B. Humanalbumin, Fresh-frozen-Plasma) ersetzt.
→ Klinisch-relevant: Eine hochdosierte Glukokortikoid-Gabe ist nur bei der chronischen Form wirksam und indiziert.
→ Prognose:
→ I: Die klinische Erholung erfolgt immer in umgekehrter Reihenfolge der neurologischen Ausfälle, zumeist über Wochen bis Monate. Es kann zu motorischen Schwächen und Reflexdefiziten kommen, ohne dass das tägliche Leben beeinträchtigt ist. In 5-20% der Fälle bleiben neurologische Defizite bestehen; die Gesamtmortalität liegt bei 2-3%.
→ II: Prognostisch ungünstig sind insbesondere:
→ 1) Alter > dem 60. Lebensjahr.
→ 2) Vorangegangene Infektion mit Campylobacter jejuni.
→ 3) Nachweis eines erhöhten GM1-Antikörper-Titer.
→ 4) Elektromyographischer Nachweis einer ausgeprägten axonalen Schädigung (nach 2-3 Wochen).