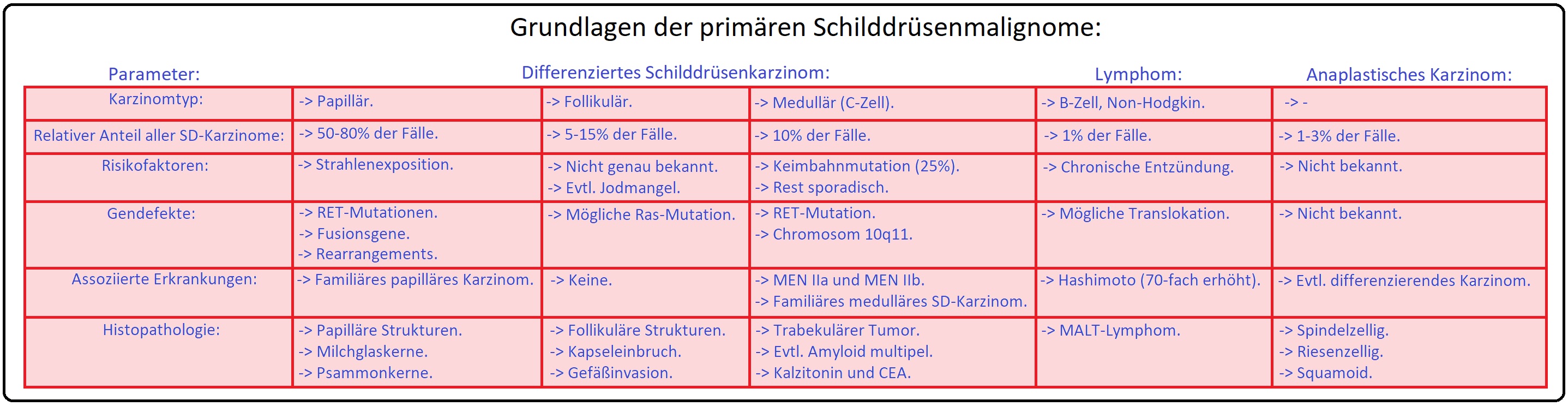
→ Definition: Das differenzierte Schilddrüsenkarzinom ist definiert als eine maligne, epitheliale Neoplasie, welche von den Thyreozyten der Schilddrüse ausgeht. Hierbei wird zwischen 2 Subtypen, dem papillären und follikulären Karzinom, unterschieden.
→ Epidemiologie:
→ I: Das Schilddrüsenkarzinom ist die häufigste endokrine Neoplasie mit einer Inzidenz von 4/100000/Jahr (die Inzidenz hat insbesondere in der letzten Dekade weltweit deutlich zugenommen). Der Manifestationsgipfel liegt jenseits des 50. Lebensjahrs.
→ II: Bei der differenzierten Form sind Frauen 3 x häufiger als Männer betroffen.
→ 1) Follikuläres SD-Karzinom: (35% der Fälle) Vermehrtes Auftreten in Jodmangelgebieten mit hoher Struma-Inzidenz. Die Häufigkeit nimmt mit dem Alter deutlich zu.
→ 2) Papilläres SD-Karzinom: (50% der Fälle) Häufig in Gebieten mit normaler Jodversorgung. Kann in jedem Alter auftreten; betrifft vermehrt jüngere Patienten
→ III: Beim anaplastischen - und medullären Schilddrüsenkarzinom ist die Geschlechterverteilung gleich.
→ Ätiologie: Die Pathogenese der differenzierten Schilddrüsenkarzinome ist nicht genau bekannt, jedoch erhöhen ionisierende Strahlungen das Erkrankungsrisiko drastisch. Risikofaktoren für die Entwicklung einer Maglinität aus einem Knoten sind nach der American-Association of Clinical Endocrinologists (= AACE) u.a. Bestrahlung des Kopfes oder Kehlkopfbestrahlung in der Vorgeschichte, positive Familienanamnese mit einem medullären Schilddrüsenkarzinom, eine MEN 2, ein fixierter derber Knoten, der ein schnelles Wachstum aufweist, männliches Geschlecht sowie Alter < 20. Lebensjahr oder > 70. Lebensjahr und nicht zuletzt Symptome wie vergrößerte Lymphknoten, Heiserkeit, Schmerzen, Dysphagie und Dyspnoe, etc. Weitere prädisponierende Faktoren sind insbesondere genetische Syndrome wie die familiäre Polyposis, das Cowden-Syndrom, Werner-Syndrom, etc.
→ Pathogenese: Im Vordergrund stehten zahlreiche molekulargenetische Veränderungen im Bereich der Onkogene und Tumorsupressorgene:
→ I: Follikuläres Schilddrüsenkarzinom: Sowohl beim follikulären Adenom als auch beim Karzinom lassen sich in 20-30% der Fälle Mutationen in der ras-Protoonkogenfamilie (H-ras, K-ras und N-ras) eruieren. Es wird in diesem Zusammenhang angenommen, dass das follikuläre Adenom ein frühes Phänomen der Tumorgenese darstellt.
→ II: Papilläres Schilddrüsenkarzinom: Im Mittelpunkt der Pathogenese steht die Aktivierung RET-PTC-Onkogens aufgrund einer Translokation des RET-Protoonkogens und des H4-Gens. Weitere molekulare Veränderungen des papillären Schilddrüsenkarzinoms wie trk-Rearrangement, Überexpression von ras-Onkogen-Produkten, etc. sind weniger spezifisch, weisen jedoch auf einen besonders aggressiven Charakter und eine rapide Tumorprogression hin.
→ Pathohistologie:
→ I: Papilläres Schilddrüsenkarzinom: Pathomorphologisch lassen sich beim papillären Schilddrüsenkarzinom verschiedene Varianten unterscheiden:
→ 1) Papilläres Mikrokarzinom: Nach der WHO werden alle papillären SD-Karzinome, deren maximaler Durchmesser < 1cm ist, als Mirkokarzinome bezeichnet, unabhängig davon, ob der Tumor gekapselt ist oder nicht.
→ 2) Gekapseltes papilläres Karzinom: Dieser Karzinomtyp macht etwa 10% aller papillären Schilddrüsenkarzinome aus und wird komplett von einer Faserkapsel ummantelt.
→ 3) Follikuläre Variante: Des papillären Schilddrüsenkarzinom. Diese Form weist ein fast reines follikuläres Wachtumsmuster auf, und es bedarf beim Nachweis papillärer Strukturen (charakteristische Kernveränderungen sowie bevorzugte lymphogene Metastasierung) größter Sorgfalt.
→ 4) Weitere Typen: Weitere pathomorphologische Varianten des papillären Schilddrüsenkarzinoms sind u.a.:

→ II: Follikuläres Schilddrüsenkarzinom: Diese maligne Neoplasie hat ihren Ursprung im Follikelepithel der Schilddrüse und besitzt pathomorphologisch auch verschiedene Varianten, die sich z.T. in ihrer Prognose stark differenzieren.

→ Klassifikation: Pathohistologische Einteilung der Schilddrüsenkarzinome:
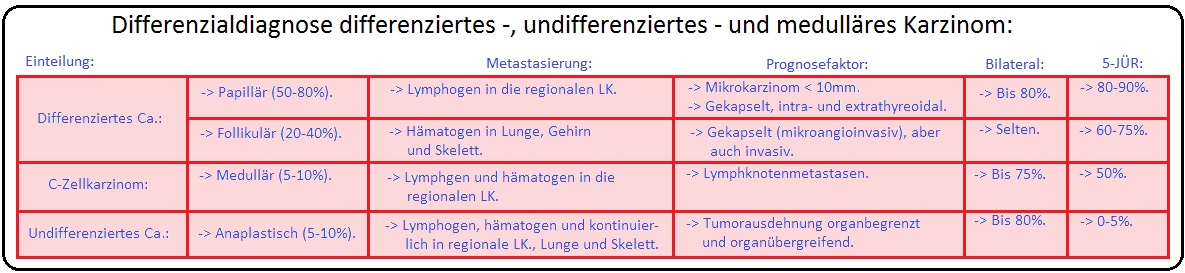
→ I: Differenziertes SD-Ca:
→ 1) Follikuläres SD-Ca: Das follikuläre SD-Karzinom macht 35% aller Malignome der SD aus und stellt somit die 2.-häufigste Karzinomart dar; es handelt sich zumeist um ein solitäres Karzinom und metastasiert frühzeitig hämatogen in Knochen und Lunge.
→ 2) Papilläres SD-Ca: Ist mit 50% das häufigste SD-Karzinom und metastasiert vorwiegend lymphogen in die regionären Lymphknoten, z.T. bevor der Primärtumor nachweisbar ist (Mikrokarzinom < 1cm, das lange Zeit asymptomatisch bleibt). Der Tumor tritt überwiegend multifokal auf und lässt sich gut mit einer 131Jod-Therapie behandeln.

→ II: Undifferenziertes SD-Ca: Das anaplastisches Schilddrüsenkarzinom stellt mit 5% der Fälle eine sehr seltene Erkrankung dar. Zudem nimmt es nicht am Jodumsatz teil, sodass eine 131Jod-Therapie ungeeignet ist.
→ III: C-Zell-Karzinom: (5%) Ist eine von den medullären C-Zellen ausgehende Neoplasie, die vermehrt Calcitonin produziert (Rezidivindikator).
→ Klinik: Lange Zeit asymptomatisch; erst im fortgeschrittenen Stadium manifestiert sich eine Symptomatik aufgrund eines infiltrativen Wachstums in die Nachbarorgane wie Trachea, Ösophagus, sowie Nervenbeteiligung. Klinische Zeichen sind u.a.:
→ I: Lokale Zeichen: Derber, höckriger Knoten, fixierte Haut, geschwollene nicht-schmerzhafte Lk im zervikalen und supraklavikulären Bereich.
→ II: Globusgefühl und Dysphagie,
→ III: Heiserkeit durch eine Recurrensparese,
→ IV: Inspiratorischer Stridor und Dyspnoe bei Befall der Trachea.
→ V: Horner-Syndrom: Selten, mit Miosis, Ptosis und Endophthalmus.
→ VI: Fernmetastasen betreffen besonders die Lunge und das Skelettsystem.
→ Diagnose:
→ I: Die Diagnose wird häufig bei der Abklärung einer nodulären Struma gestellt.
→ II: Bestimmung des Thyreoglobulins (Tumormarker bei Rezidivabklärung, evtl. auch TPA = tissue-polypeptid-antigen)
→ III: Sonographie: Besonders verdächtig sind unregelmäßig begrenzte, echoarme Areale mit Mikrokalzifikationen. Weitere sonographische Malignitätsbefunde sind u.a.:
→ 1) Kein Halo,
→ 2) Intranoduläre Vaskularisierung etc.
→ IV: Szintigraphie: Kalte Knoten, die nicht speichern.
→ V: Fernmetastasen: Zum Ausschluss Röntgen-Thorax, CT, Sono-Abdomen, Knochen-Szintigraphie.

→ Klinisch-relevant: Ein szintigraphisch kalter SD-Knoten, der sonographisch nicht echofrei ist, ist immer tumorverdächtig. Folgende Untersuchungen sind hierbei obligat.
→ A) Feinnadelbiospie mit Aspirationszytologie ist in 90% wegweisend.
→ B) Bei negativem Befund und fortbestehendem Tumorverdacht; operative Tumorentfernung und nachfolgende histologische Untersuchung.
→ Differenzialdiagnose: Vom differenzierten Schilddrüsenkarzinom sind u.a. nachfolgende Erkrankungen abzugrenzen:
→ I: Zystische Schilddrüsenveränderungen: Einschließlich der Zystenblutungen.
→ II: Thyreoditis: Wie die Hashimoto-Thyreoiditis oder die subakute Thyreoiditis der Quervain.
→ III: Struma nodosa: (euthyreote Struma) Mit benignen, regressiven Knoten; die Abgrenzung ist z.T. sehr schwierig, sodass eine Thyreoidektomie erfolgen muss.
→ IV: Weitere Differenzialdiagnosen: Sind insbesondere:
→ 1) Lymphom der Schilddrüse sowie
→ 2) Metastasen extrathyreoidaler Malignome.
→ Therapie: Die genaue histologische Bestimmung des differenzierten Schilddrüsenkarzinoms für die Behandlung von besonderer Bedeutung.
→ I: Operative Therapie:
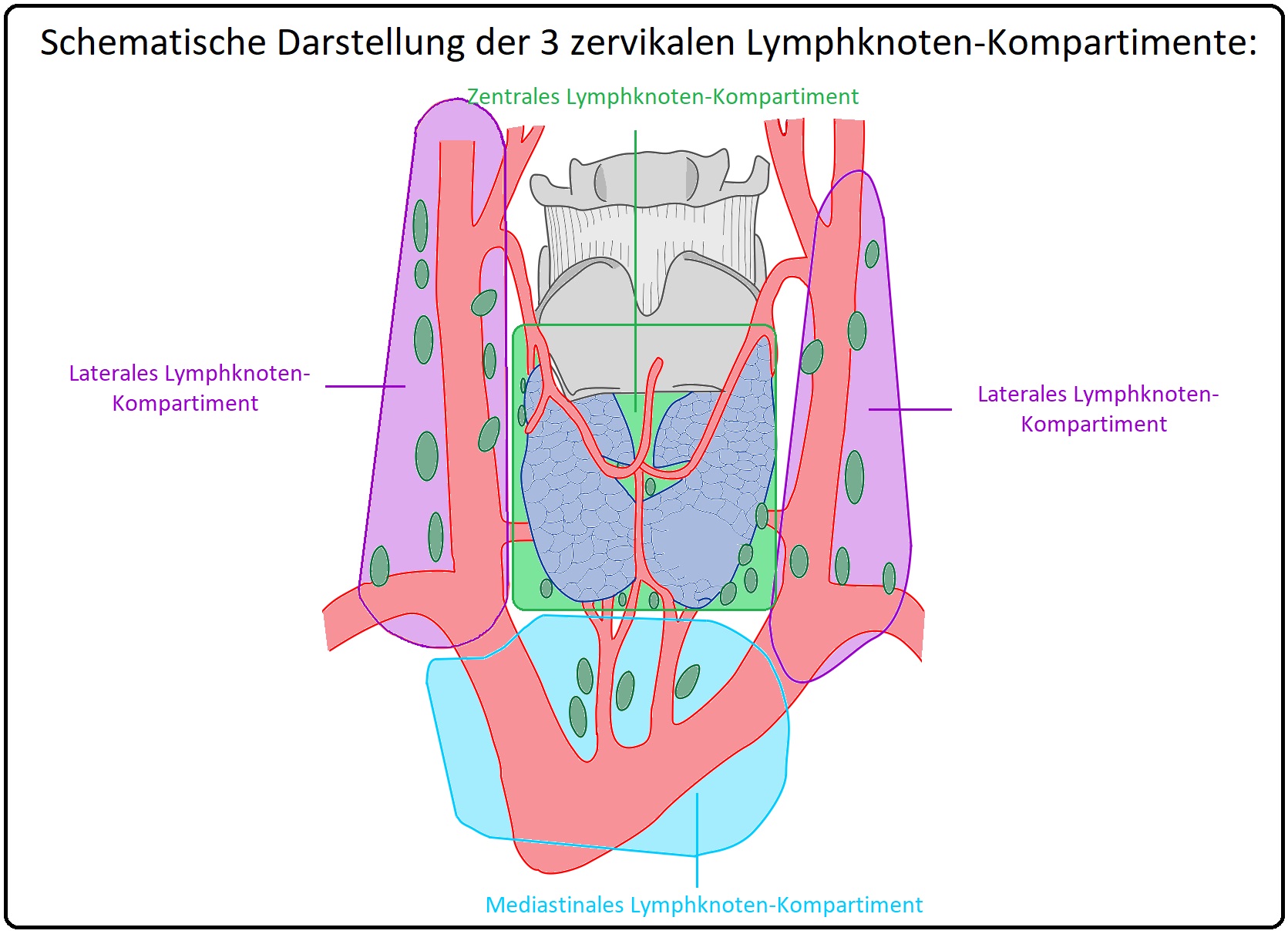
→ 1) In den meisten Fällen ist eine radikale totale Thyreoidektomie mit Entfernung der Lymphknoten im zentralen Kompartiment indiziert. Der gleichzeitige, metastatische Befall der regionären Lymphknoten führt zu einer zusätzlichen chirurgischen Neck-Dissektion.
→ 2) Bei einem solitären, papillären Mikrokarzinom ohne Nachweis von Lymphknotenmetastasen kann die Hemithyreoidektomie eine kurative Therapie sein.
→ II: Ablative Radiojodtherapie: In der 3.-4. postoperativen Woche erfolgt ein 131Jod-Ganzkörperscan zum Ausschluss/Nachweis von Thyreozytenresten und Metastasen; sind Metastasen eruierbar, sollte eine hochdosierte Radiojodtherapie in mehreren Fraktionen, bis kein Jod-speicherndes Gewebe mehr nachweisbar ist, durchgeführt werden.
→ III: Weitere Therapiemaßnahmen:
→ 1) Im Anschluss (Radiojodtherapie) ist eine hochdosierte Hormonlangzeittherapie mit Levothyroxin (150-300µg/d) zur Suppression von TSH, (um die wachstumsfördernde Wirkung von TSH auf evtl. Metastasen zu unterbinden) indiziert.
→ 2) Evtl. externe Radiatio als pallitative Therapie zur Reduktion von Tumorkomplikationen z.B. bei vehementen Schmerzen.
→ IV: Nachsorge: Kontrolluntersuchungen im Abstand von 6 Monaten sind obligat mit:
→ 1) Bestimmung des Thyreoglobulins (TG). Dieses wird vom normalen SD-Gewebe sowie von den Zellen des SD-Karzinoms gebildet. Durch die radikale Thyreoidektomie spricht ein Anstieg des TG für ein Tumorrezidiv bzw. Metastasen.
→ 2) Evtl. Thyreogobulin-Bestimmung nach Gabe von rekombinantem humanem TSH (rhTSH) zum Nachweis von Tumorrezidiven/Metastasen.
→ 3) Bei Verdacht auf ein Rezidiv bzw. eine Metastase sollte immer ein 131Jod-Ganzkörperscan durchgeführt werden.
→ 4) Weitere Nachweisoptionen von Metastasen sind Röntgen-Thorax, CT, Ganzkörperskelett-Szintigraphie etc.
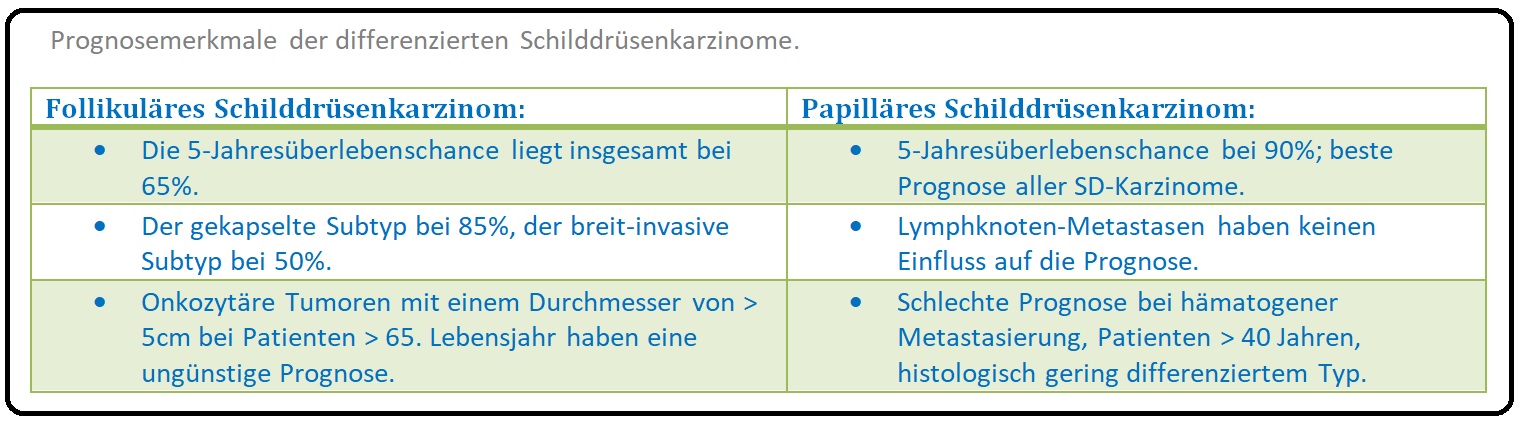
→ Prognose: Bei adäquater Therapie liegt die 5-Jahresüberlebenschance beim papillären Schilddrüsenkarzinom bei > 90% und hat von allen SD-Karzinomen die beste Prognose. Beim follikulären SD-Karzinom liegt sie bei 65%. Weitere wichtige Prognosefaktoren sind u.a. Tumorgrößer, histologischer Typ, Ausmaß der extrathyreoidalen Invasion, Lymphknoten- und Fernmetastasen sowie das Lebensalter.