→ Definition: Bei den gastrointestinalen Stromatumoren handelt es sich um mesenchymale Tumoren des GIT, die von den nicht-neuronalen gastrointestinalen Schrittmacherzellen, den sogenannten Cajalzellen, ausgehen. Sie weisen sowohl neuronale - als auch den glatten Muskelzellen zugehörige Charakteristika auf. Ein wichtiger immunhistochemischer Marker dieser Stromatumoren ist die Expression des c-kit-Protoonkogens (= membranständige Rezeptor-Tyrosinkinase CD117).
→ Epidemiologie:
→ I: Die gastrointestinalen Stromatumoren stellen mit 10-15 Neuerkrankungen/1000000 Einwohner pro Jahr (= 800-1200 Neuerkrankungen pro Jahr) eine seltene Erkrankung dar.
→ II: Es zeigt sich ein überwiegend sporadisches Auftreten mit einem Manifestationsgipfel zwischen dem 55.-65. Lebensjahr, wobei Männer etwas häufiger als Frauen betroffen sind.
→ Ätiopathogenese:
→ I: Ursprung der GIST sind die interstitiellen Zellen von Cajal:
→ 1) Durch Expression der aktivierten Mutation des c-KIT-(CD117-) Protoonkogens, das die Typ-III-Rezeptor-Tyrosinkinase kodiert oder
→ 2) Mutation des PDGFR-Alpha-Protoonkogens (= Platelet-derived-Growth-Factor-Rezeptor-Alpha), das wiederum eine weitere Rezeptor-Tyrosinkinase codiert.
→ II: Im c-KIT-Gen ist am häufigsten das Exon 11 mutiert, gefolgt vom Exon 9, Exon 13 und Exon 17. Beim PDGFR-Alpha-Gen ist wiederum am häufigsten das Exon 18 mutiert gefolgt von Exon 12 und Exon 14.
→ III: Selten entsteht der gastrointestinale Stromatumor aus einer inaktivierenden Mutation des Succinat-Dehydrogenase-Komplexes; dies betrifft vor allem die GIST des Kindesalters mit Deletion des Exon 11; es existieren aber auch Punktmutationen.
→ IV: Morphologie: Die gastrointestinalen Stromatumoren sind stark vaskularisierte in der Submucosa lokalisierte Tumoren, die charakteristischerweise nach außen wachsen (weg vom gastrointestinalen Lumen).
→ V: Weitere Risikofaktoren für die Entwicklung eines GIST sind u.a.:
→ 1) Genetische Disposition mit familiärer Häufung aufgrund einer autosomal-dominant vererbten KIT-Mutation, die zu multiplen gastrointestinalen Stromatumoren.
→ 2) Bestimmte Vorerkrankungen wie das Carney-Stratakis-Syndrom oder Carney-Triad-Syndrom sowie die Neurofibromatose Typ I gehen mit einem erhöhten GIST-Risiko einher.

→ Klassifikation: Die gastrointestinalen Stromatumoren lassen sich unterteilen:
→ I: Nach der Lokalisation:
→ 1) Die häufigste Lokalisation des GIST ist der Magen (50%-60% der Fälle, sind zumeist benigne) gefolgt vom Dünndarm (20%-30% der Fälle, weisen zumeist eine Zwischenstellung auf).
→ 2) Seltener sind die Tumoren im Bereich des Ösophagus (2-5%, häufig maligne), Ampulla Vateri, Kolon (zumeist maligne), Rektum (5-12%), Appendix und Anus. Auch extragastrointestinale GIST (z.B. ins Omentum oder Mesenterium) sind möglich, aber sehr selten).
→ II: Nach der Histologie:

→ Klinik: Die klinische Symptomatik der gastrointestinalen Stromatumoren sind sehr unspezifisch und insbesondere von der Lokalisation abhängig. Nicht
selten ist der GIST ein Zufallsbefund; größere Tumoren können Obstruktionen und weitere Symptome hervorrufen.
→ I: Mögliche Beschwerden sind Völlegefühl, unspezifische abdominale Schmerzen, Subileus-Symptomatik.
→ II: Dysphagie, Übelkeit und Gewichtsverlust (Tumorkachexie), aber auch palpable Tumor im fortgeschrittenen Stadium mit möglicher Bauchumfangsvermehrung.
→ III: Weitere mögliche Symptome sind insbesondere gastrointestinale Blutungen, Anämie und eine Verschlechterung des Allgemeinzustandes.
→ Metastasierung: In 20-50% der Fälle weisen die Patienten mit GIST bereits zu Diagnosestellung eine Metastasierung auf.
→ I: Fernmetastasen manifestieren sich sich insbesondere in Leber und Peritoneum (Omentum). Extraabdominale Metastasen (liegen in < 10% der Fälle vor) z.B. Lungenmetastasen sind nur sehr selten nachweisbar und sprechen für ein fortgeschrittenes Stadium.
→ II: Lymphknoten sind sehr selten sodass eine Lymphknotendissektion insbesondere der regionären Lymphknoten nicht empfohlen wird.
→ Diagnose: Häufig handelt es sich bei den gastrointestinalen Stromatumoren um Zufallsdiagnosen im Rahmen von Untersuchungen oder Therapien. Diagnostisch spielen insbesondere radiologische (CT, PET) und endoskopische Verfahren (einschließlich Endosonographie) die wichtigste Rolle.
→ I: GIST-Tumoren imponieren häufig als gut abgrenzbare polyzyklische Tumoren mit Verdickung der Lamina und zentralen zystischen Bereichen.
→ II: CT:
→ 1) In der kontrastmittelverstärkten CT weisen vor allem größere gastrointestinale Stromatumoren ein Kontrastmittel-Enhancement der Randbereiche auf; sie sind beweisend für Hypervaskularisation sowie mögliche Einblutungen und Nekrosen. Des Weiteren dient die CT-Untersuchung dem Staging und der OP-Planung.
→ 2) Die PET-CT-Untersuchung ermöglicht einerseits morphologische, funktionelle und metabolische Auskunft zu geben, anderseits hat es in der Beurteilung des therapeutischen Ansprechen auf Tyrosinkinase-Inhibitoren große Bedeutung erlangt.
→ III: Insbesondere in der Ausbreitung des Tumor hat sich die Endosonographie einen hohen Stellenwert.
→ IV: Endgültige Klarheit für die Diagnose der gastrointestinalen Stromatumoren ist die histologische Untersuchung des Gewebes mittels z.B. Feinnadelbiopsie oder einer anderen Gewebeentnahme.

→ V: Eine molekulargenetische Untersuchung zur Bestimmung des c-KIT- oder PDGFR-Alpha Mutationsstatus ist insbesondere von therapeutischer Relevanz.
→ Klinisch-relevant: Diagnosekriterien des GIST sind Tumorlokalisation, Tumorgröße, Mitosezahl, Nachweis von c-KIT-Mutationen. Intestinale Tumoren > 5cm mit hoher Mitosezahl sind in der Regel maligne.
→ Differenzialdiagnose: Von den gastrointestinalen Stromatumoren müssen insbesondere nachfolgende Erkrankungen abgegrenzt werden:
→ I: Ulkus ventriculi, Ulkus duodeni oder Ulcus pepticum jejuni.
→ II: Malignome wie das Magenfrühkarzinom, Magenkarzinom, MALT-Lymphom, aber auch kolorektales Karzinom, etc.
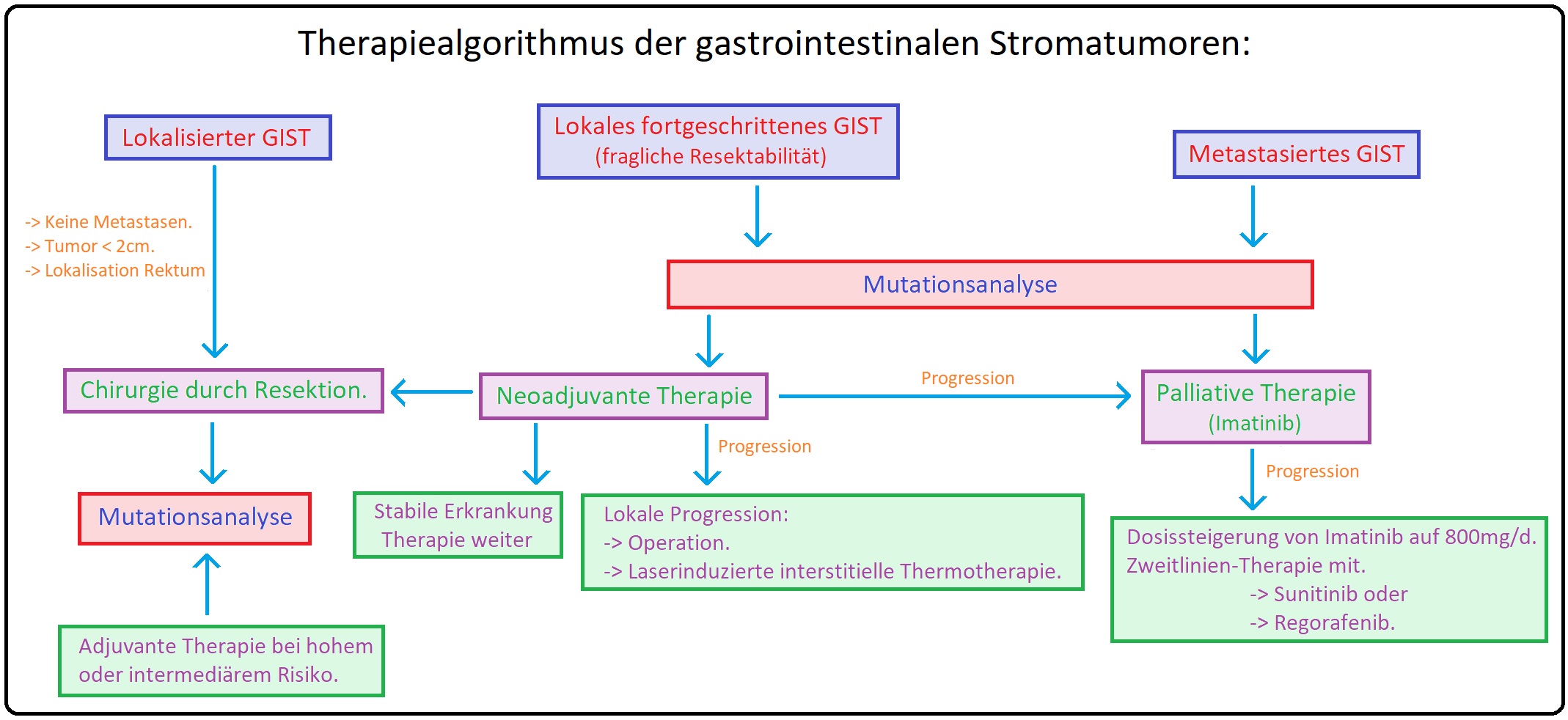
→ Therapie: Die Therapie des gastrointestinalen Stromatumors besteht auf 2 Säulen, der operative - und medikamentösen Therapie. Hierbei muss die Indikation sorgfältig und interdisziplinär getroffen werden.
→ I: Chirurgische Therapie: Alle lokalen gastrointestinalen Stromatumoren mit einer Größe < 2cm müssen operativ reseziert werden. Hierbei ist primäres Ziel den Tumor in toto zu entfernen (= vollständige Tumorresektion einschließlich der Tumorkapsel), um das Rezidivrisiko zu minimieren.
→ 1) Eine routinemäßige Lymphadenektomie wird generell nicht empfohlen.
→ 2) Die durchschnittliche Rezidivfreiheit bei R0-Resektion beträgt 18-24 Monate.
→ 3) Ist eine R0-Resektion nicht möglich, wird eine präoperative neoadjuvante Therapie zur Tumorverkleinerung mit dem Tyrosinkinaseinhibitor, Imatinib, empfohlen. Unter der Imatinib-Therapie wird in 3-4monatigen Intervallen die Resektabilität des Tumors überprüft. Bei Erreichen der Remission ist die Resektion indiziert (zumeist nach 6-12 Monaten).
→ Klinisch-relevant: Beim chirurgischen Eingriff ist immer eine intraoperative Tumorruptur zu vermeiden, da sich eine intraperitoneale Sarkomatose negativ
auf das Gesamtüberleben auswirkt.
→ II: Medikamentöse Therapie: Mittel der Wahl ist der Tyrosinkinase-Inhibitor, Imatinib. Hierbei handelt es sich um ein oral applizierbares Phenylaminopyrimidinderivat, das mit ATP um die spezifischen Andenosinbindungsstelle konkurriert und somit die für die Rezeptoraktivierung notwendige Phosphorylierung hemmt. Hierdurch werden die intrazellulären Kaskaden, die zum Tumorwachstum führen, unterbrochen. Indikationen sind u.a.:
→ 1) Seit 2002 ist Imatinib in einer Dosierung von 400mg/d insbesondere für die metastasierten gastrointestinalen Stromatumoren zugelassen.
→ 2) Im Anschluss an eine operative Intervention ist eine adjuvante Therapie mit Imatinib 400mg/d über 3 Jahre substituiert.
→ 3) Ausnahme bilden die c-KIT-Exon 9 Mutation und PDGFR-Alpha-D842V-Mutation, da sie Imatinib-resistent sind. Hierfür wird die Zweitlinientherapie mit Sunitinib empfohlen.
→ Klinisch-relevant: Die gastrointestinalen Stromatumoren sprechen nicht auf Chemo- und Strahlentherapie an und haben somit keinen Stellenwert.
→ III: Nachsorge: Bei niedrigem Risiko halbjährliche, bei intermediärem oder hohem Risiko 3-4 monatige Intervalle in den ersten 3 Jahren, danach alle 6 Monate und schließlich nach 5 Jahren jährliche Tumorkontrollen. Im Mittelpunkt der Nachsorge stehen die klinische Untersuchung sowie CT von Abdomen und Becken.
→ Prognose: Die 5-Jahresüberlebenschance beim gastrointestinalen Stromatumor ist sehr variabel, reicht von 13%-90% und ist im Wesentlichen von der
Risikoeinschätzung (Tumorgröße, Tumorlokalisation Mitoserate) abhängig.
→ I: Die mediane Überlebenszeit bei metastasierter Erkrankung liegt derzeit bei 60 Monaten, die 5-Jahresüberlebensrate wiederum bei 45%.
→ II: Insbesondere Patienten mit einer c-KIT-Exon-11-Deletion haben ein hohes Rezivrisiko.
→ III: Kleine GIST des Magens (<2cm) haben nach einer Resektion nur ein geringes Rezidivrisiko, sodass (abhängig vom Alter, Komorbidität, perioperativen Risiken) eine 5-6monatige Verlaufskontrolle empfohlen wird.
→ IV: Bei kleinen GIST anderer Lokalisation (insbesondere Rektum) wird aufgrund der hohen Metastasierungs- und Rezidivrate eine engmaschige Verlaufskontrolle im Abstand von 3 Monaten empfohlen.