→ Definition: Bei der Hämophilie handelt es sich um eine (geschlechtsgebundene) X-chromosomal-rezessiv vererbte Gerinnungsstörung aufgrund einer Aktivitätsstörung der Gerinnungsfaktoren VIII (= Hämophilie A) und IX (= Hämophilie B) mit Blutungserscheinungen unterschiedlichen Ausmaßes.
→ Klinisch-relevant: Andere Gerinnungsdefekte, die u.a. die Faktoren II, V, VII, IX und X betreffen, können ein ähnliches klinisches Erscheinungsbild verursachen, werden jedoch nicht zur Hämophilie gezählt.
→ Epidemiologie:
→ I: Die Prävalenz der Hämophilie liegt in Deutschland bei 1/10000 Personen, wobei 80% der Fälle an Hämophilie A und nur 20% an der Hämophilie B leiden.
→ II: Wegen der X-chromosomal-rezessiven Vererbung erkranken Männer (XY) immer, während Frauen (XX) zumeist Konduktorinnen sind und nur sehr selten das charakteristische klinische Bild aufweisen.
→ III: Bei 30% (-50%) der Fälle zeigt sich eine Spontanmutation mit unauffälliger Familienanamnese.
→ Ätiopathogenese: Durch den angeborenen Mangel an Faktor VIII und IX-Aktivität wird das intrinsische plasmatische Gerinnungssystem gestört.
→ I: Hämophilie A: Manifestiert sich infolge eines angeborenen Faktor VIII Mangels. Das Faktor-VIII-Molekül ist ein aus einer leichten - und einer schweren Kette bestehendes Dimer. Es enthält 3A-, 2C- und eine dazwischenliegende inaktive B-Domäne. Die schwere Kette besteht aus 2A und der B-Domäne. Die leichte Kette aus der 3.A- und den zwei C-Domänen.
→ 1) Das Faktor-VIII-Gen liegt auf dem langen Arm des X-Chromosoms und weist eine Vielzahl von Defekten auf, die zu einer Hämophilie A führen können. Insbesondere bei schwerer Hämophilie zeigen sich u.a. große Gendeletionen, Nonsense-Mutationen sowie (relativ häufig) eine Inversion auf dem Intron 22 (Missense-Mutationen an funktionellen Stellen führen zumeist zur leichteren Hämophilie).
→ 2) Faktor VIII wird hauptsächlich in den Hepatozyten synthetisiert.
→ 3) Beim Faktor VIIIa handelt es sich um einen Kofaktor für die Aktivierung von Faktor X mit Hilfe von Faktor IXa in Gegenwart von Kalzium und Phospholipiden. Faktor VIII wiederum wird durch Thrombin aktiviert.
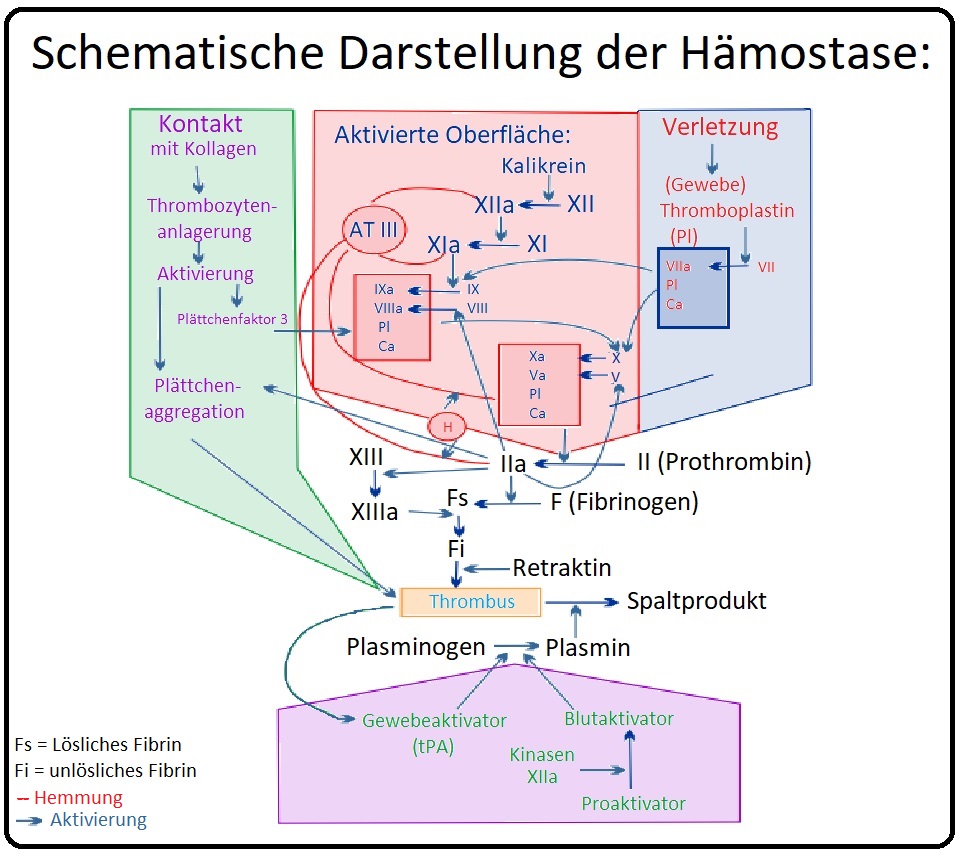
→ II: Hämophilie B: Bei der Hämophilie B besteht ein Mangel an Faktor IX.
→ 1) Beim Faktor IX (Faktor-IX-Gen liegt auch auf dem langen Arm des X-Chromosoms) handelt es sich um ein einkettiges Glykoprotein, das in der Leber Vitamin-K-abhängig synthetisiert wird.
→ 2) Das Molekül besitzt u.a.:

→ 3) Der Faktor IX wird insbesondere durch den Faktor VIIa (weniger des Faktor XIa) in Gegenwart von Kalzium und Phospholipiden aktiviert. Der Faktor IXa wiederum aktiviert in Anwesenheit von Phospholipiden, Kalzium und Faktor VIIIa den Faktor X (X→Xa).
→ III: Pathogenese: Durch den Faktor-Mangel kommt es zu einer verminderten Aktivität des intrinsischen plasmatischen Gerinnungssystems mit konsekutiv reduzierter Aktivierung von Faktor X und Fibrin. Folge ist eine verlangsamte Fibrinbildung und da Thrombin auch den Faktor XIII aktiviert eine gestörte Quervernetzung von Fibrin.
→ Klinik: Das klinische Bild der beiden Hämophilie- Formen ist nicht voneinander zu unterscheiden (vielmehr erfolgt dies durch die laborchemische Bestimmung der Faktoren).
→ I: Erste Hinweise für eine Hämophilie sind ausgedehnte Blutungen nach Bagatelltraumen, spontane Einblutungen in Muskel und Gelenke (Hauptlokalisationen sind Ellenbogen- und Kniegelenk, seltener Schultergelenk), aber auch postoperative Nachblutungen (sowohl Sofortblutungen, als auch besonders charakteristisch Spätblutungen nach Stunden und Tagen).
→ II: Weitere Symptome umfassen schmerzlose Hämaturie (später nach Bildungen von Gerinnseln können sich Koliken entwickeln) und große lokalisierte Hämatome.
→ III: Rezidivierende Gelenk- und Muskelblutungen (insbesondere im Bereich des Unterarm und der Unterschenkelmuskulatur, seltener die Oberschenkelmuskulatur betreffend) führen nicht selten zu sekundären Folgeschädigungen wie u.a.
→ 1) Muskelatrophie und Muskelkontraktur.
→ 2) Chronische Synovitis und Schädigungen des Bandapparates sowie Gelenkversteifung.
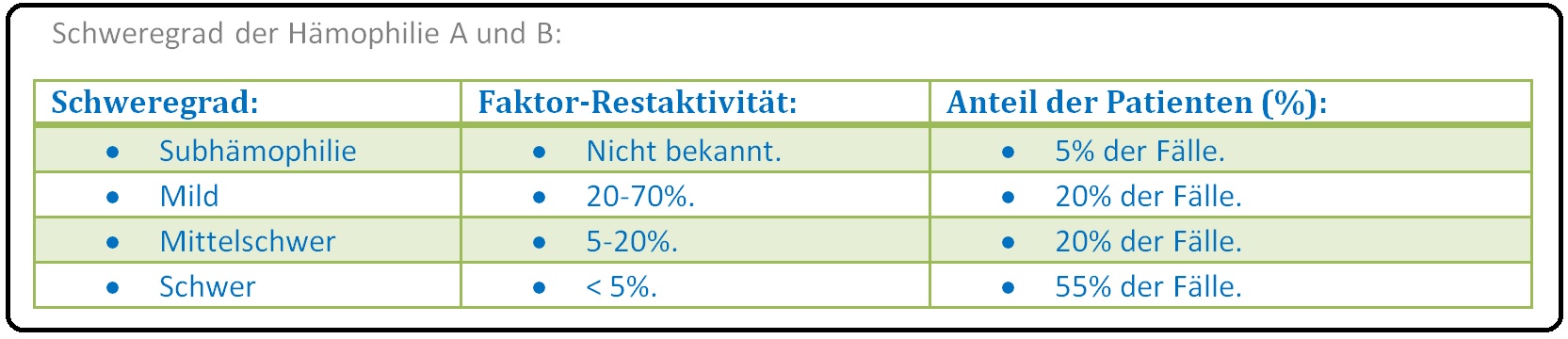
→ IV: Subhämophile Patienten mit milder Verlaufsform entwickeln Blutungen nur nach Traumen und größeren Operationen oder Verletzungen.
→ Komplikationen: Wichtige und z.T. schwerwiegende Komplikationen der Hämophilie sind u.a.:
→ I: Massive Einblutungen in die Muskulatur (Kompartmentsyndrom, hypovolämischer Schock) und das Retroperitoneum mit konsekutivem Resorptionsfieber und Leukozytose. (Differenzialdiagnose akutes Abdomen, Appendizitis, etc.)
→ II: Intrakranielle Blutungen sind in bis zu 25% der Fälle Todesursache.
→ Klassifikation: Die Hämophilie A und B lässt sich in 4 klinische Schweregrade unterteilen:
→ Diagnose: Wichtig bei der Diagnose ist die umfangreiche Eigenanamnese mit klinischer Symptomatik (vermehrtes Bluten, Nachblutungen, Gelenkblutungen) sowie die Familienanamnese (evtl. positiv).
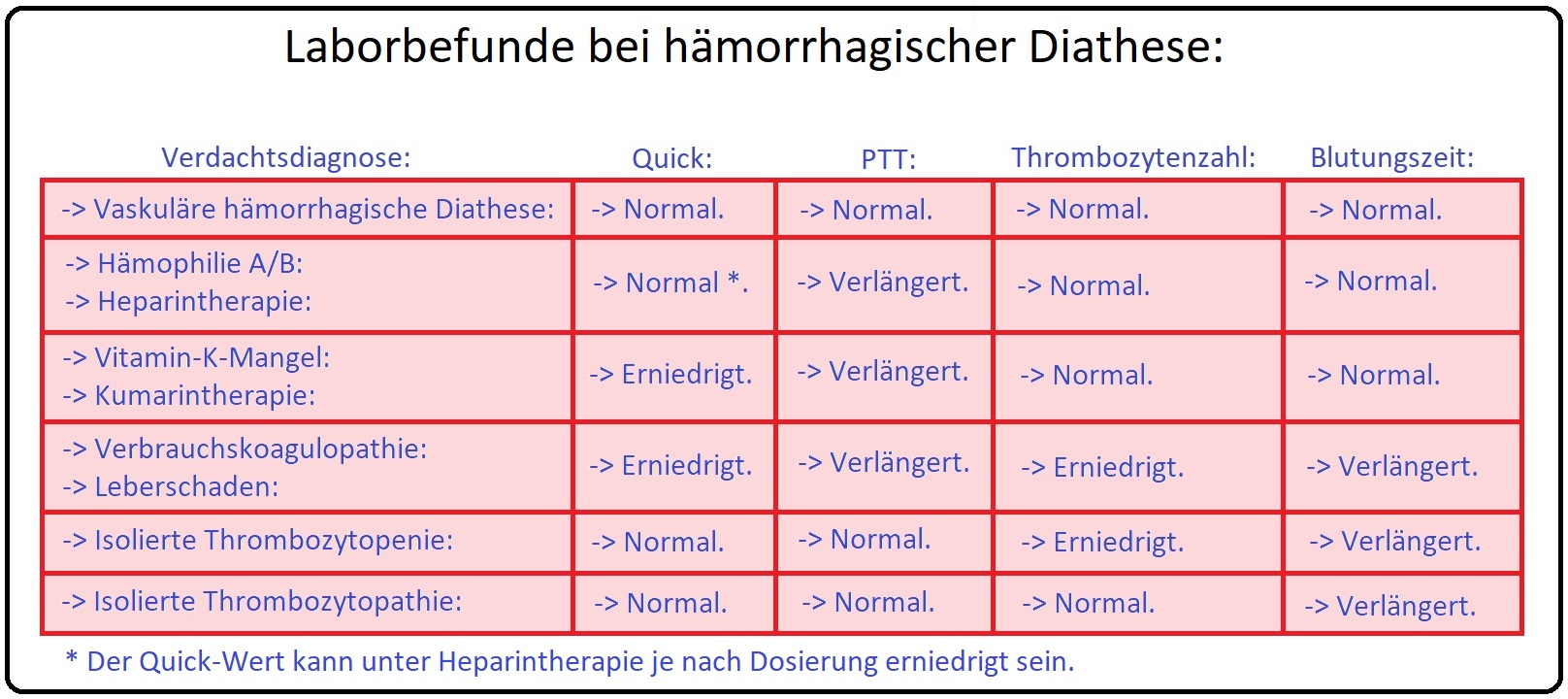
→ I: Laborchemisch ist die verlängerte aPTT richtungsweisend; die primäre Blutstillung bzw. Blutungszeit sowie die Thombin- bzw. Prothombinzeit sind regelrecht (Abb: Wichtige Gerinnungsparameter und ihre Normwerte).

→ II: Zur Sicherung der Diagnosestellung ist die Bestimmung der Faktor VIII- und Faktor IX-Aktivität obligat.
→ III: Schließlich sollte ein kompletter Gerinnungs- und Thrombozytenstatus zur Erfassung der Hämostase-Funktion erfolgen.
→ Differenzialdiagnose: Von der Hämophilie A/B müssen insbesondere nachfolgende Erkrankungen abgegrenzt werden:
→ I: Von-Willebrand-Jürgens-Syndrom.
→ II: Antikörper-bedingter Faktor-VIII-Mangel (= Hemmkörperhämophilie) grenzt sich insbesondere in der Anamnese und dem Nachweis eines Antikörper gegen Faktor VIII ab.
→ Therapie: Die Behandlung der Hämophilie erfolgt primär symptomatisch.
→ I: Allgemeinmaßnahmen: Umfassen u.a. bei äußeren Blutungen Anlage eines Druckverbandes, bei Gelenkblutungen Ruhigstellung, Hochlagerung, Kühlung, etc.
→ II: Prophylaxe: Wichtige Prophylaxen bei Hämophilie-Patienten sind insbesondere:
→ 1) Keine intramuskuläre Injektionen.
→ 2) Keine Applikation von Thrombozytenaggregationshemmern (z.B. ASS), HAES oder Dextranen, etc.
→ III: Substitutionstherapie: Die Applikation von Faktorenkonzentrat ist Mittel der ersten Wahl bei schweren Formen:
→ 1) In der Akuttherapie bei Blutungen ist die sofortige intravenöse Gabe von 25-50IE/kgKG sowie ggf. eine lokale Blutstillung indiziert.
→ Klinisch-relevant: Zur Berechnung der Dosis existiert eine Faustregel; sie besagt, dass eine Einheit Faktor-VIII- oder IX-Konzentrat die Aktivität im Blut um 1% pro kgKG anhebt. Entsprechend berechnet sich die Gesamtmenge, um den Wert z.B. von 50 oder 70% der Norm zu erhalten.

→ 2) Zur Dauertherapie werden bei Hämophilie A 25IE Faktor-VIII-Konzentrat 3x/Woche und bei Hämophilie B 25IE-Faktor IX-Konzentrat 2x/Woche verabreicht.
→ IV: Bei leichteren Formen der Hämophilie A ist bei kleineren Blutungen oder Operationen (z.B. Zahnextraktion) die Gabe von Desmopression (DDAVP) in einer Dosierung von 0,4µg/kgKG über 30min möglich (intravenös alle 12 Stunden über maximal 5 Tage). Bei mittelschwerer Hämophilie im Bedarfsfall z.B. im Rahmen von Blutungskomplikationen steht das Anheben der Gerinnungsfaktoraktivität im Vordergrund.
→ V: Nicht zuletzt ist bei größeren Operationen, größeren Verletzungen, etc. bei allen Formen der Hämophilie eine Faktorensubstitution erforderlich (Zielwert: Faktorenaktivität von 70% der Norm).
→ Klinisch-relevant: Bei der Applikation der Faktor-Konzentraten besteht immer die Gefahr der Entwicklung von unerwünschten Wirkungen; hierzu zählen u.a.:
→ I: Anaphylaxie.
→ II: Infektion mit Hepatitis B (C), HIV (heute nur noch sehr selten).
→ III: Nach häufiger Substitution Ausbildung von Antikörpern mit der Gefahr der Hemmkörperhämophilie.