- Details
- Geschrieben von: CF
- Kategorie: Gerinnungsstörungen
- Zugriffe: 2118
→ Definition: Bei der Hemmkörperhämophilie handelt es um eine sehr seltene, erworbene Gerinnungsstörung, die durch Bildung von Antikörpern gegen einen Gerinnungsfaktor gekennzeichnet ist. Ein bevorzugtes Zielprotein mit konsekutiver adaptiver Immunantwort ist der Gerinnungsfaktor VIII.
→ Epidemiologie:
→ I: Die Bildung von inhibitorischen Antikörpern gegen Gerinnungsfaktoren stellt mit einer Inzidenz von 1-4/1 Million Einwohner pro Jahr eine sehr seltene Erkrankung dar.
→ II: Die Inzidenz ist altersabhängig und wird bei Kleinkindern fast gar nicht beobachtet, während sie im Erwachsenenalter schon bei ca. 15% der Fälle liegt.
→ III: Es zeigen sich 2 Manifestationsgipfel:
→ 1) Ein kleinerer Peak zwischen dem 20.-30. Lebensjahr und
→ 2) Ein zweiter größerer - zwischen dem 65.-80. Lebensjahr, wobei beide Geschlechter gleichermaßen betroffen sind.
→ Ätiopathogenese:
→ I: Es kommt zur Bildung von Hemmkörpern bevorzugt gegen den Gerinnungsfaktor VIII (in 2-6% der Fäkke gegen den Gerinnungsfaktor IX).
→ 1) Bei der Synthese der Antikörper können 2 Subtypen unterschieden werden:

→ 2) Richtet sich der AK gegen funktionell strategische Epitope kommt es zur Funktionsstörung des Gerinnungsfaktors.
→ 3) Richtet sich der AK wiederum gegen nicht funktionell-relevante Epitope des Gerinnungsfaktors, ist eine reduzierte Halbwertszeit des Gerinnungsfaktors nachweisbar. Diese präzipitierenden AK induzieren eine Blutungsneigung durch Reduktion der Plasmakonzentration an Gerinnungsfaktoren.
→ II: Ätiologie:
→ 1) 10-30% der Patienten mit Hämophilie A bilden im Verlauf der Substitutionstherapie eine Hemmkörperphilie aus (2-6% der Patienten mit Hämophilie B).
→ 2) Auch bei erworbenen Hämophilie-Formen wie z.B. systemischer Lupus erythematodes, maligne Lymphome, medikamenteninduziert nach Penicillin- und Sufonamid-Gabe, Verbrennungen, Infektionen insbesondere Hepatitis B und Hepatitis C, aber auch im Zusammenhang mit einer Schwangerschaft oder idiopathisch, etc. kann sich eine Hemmkörperhämophilie entwickeln.
→ III: Eine familiäre Häufung ist im Gegensatz zur Hämophilie nicht bekannt.
→ Klinisch-relevant: Bei etwa 10% der Patienten mit systemischen Lupus erythematodes tritt das Lupus-Antikoagulanz, ein Phospholipid mit gerinnungshemmender Wirkung auf. 50% dieser Patienten entwickeln Thromboembolien.
→ Klinik: Das klinische Bild lässt sich von der Hämophilie abgrenzen:
→ I) Es dominieren Weichteilblutungen wie Muskel-, Haut- und Schleimhautblutungen, während intraartikuläre Blutungen extrem selten sind.
→ II: Hinzu kommen Organblutungen, intrazerebrale Blutungen sowie retropharyngeale Blutungen mit konsekutiver Verlegung der Atemwege.
→ III: Es zeigt sich also eine hämorrhagische Diathese, die nach Faktor-(VIII)-Substitution keine Besserung zeigt.
→ Komplikationen: Bei der Hemmkörperhämophilie kann es zu schwerwiegenden Komplikationen kommen; hierzu zählen insbesondere:
→ I: In bis zu 90% kommt es zu schweren bis lebensbedrohlichen Blutungen. Die Mortalitätrate liegt zwischen 8-22%.
→ II: Gefahr der Entwicklung eines anaphylaktischen Schocks.
→ Diagnose:
→ I: Wichtig hierbei ist eine umfangreiche Eigenanamnese inklusive der Abklärung von Vorerkrankungen, etc.
→ II: Labor: (Abb.: Weiterer Diagnose-Algorithmus bei verlängerter aPTT):
→ 1) Mit Bestimmung von aPTT (verlängert) und der Prothrombinzeit (nach Quick), die normal aber auch verlängert sein kann.

→ 2) Die Entwicklung des Hemmkörpers (= AK) bedeutet, dass nach Substitution des Konzentrats der Faktor-VIII-Anstieg und konsekutiv die blutstillende Wirkung ausbleibt. Im Mittelpunkt der Diagnose steht der Nachweis des Antikörpers mittels Plasmaaustauschversuchs sowie die quantitative Bestimmung des AK-Titers (in Bethesda-Einheit/ml). Bei der Titerbestimmung unterscheidet am abhängig von Bethesda-Einheit (> 5/< 5) zwischen 2 Typen:

→ Differenzialdiagnose: Differenzialdiagnostisch muss insbesondere auch die (isolierte) Hämophilie A/B und das Von-Willebrand-Jürgens-Syndrom abgegrenzt werden.
→ Therapie: Bei der Hemmkörperhämophilie stehen 2 Therapieoptionen im Vordergrund:
→ I: Gerinungssubstitution: Mit speziellen Gerinnungsfaktoren wie rekombinierter Faktor VIIa und FEIBA (= factor-eight-inhibitory-bypassing-activity). Bei letzterem handelt es sich um ein aktivierten Prothrombinkomplex.
→ II: Eine weitere Behandlungsintervention ist die Applikation von Glukokortikoiden.
→ III: Eine weitere untergeordnete Therapie möglichkeit ist die Plasmapherese.
→ IV: Bei allen Therapieoptionen ist die Behandlungskontrolle durch eine Faktorenanalyse obligat.
- Details
- Geschrieben von: CF
- Kategorie: Gerinnungsstörungen
- Zugriffe: 2327
→ Definition: Bei der Hämophilie handelt es sich um eine (geschlechtsgebundene) X-chromosomal-rezessiv vererbte Gerinnungsstörung aufgrund einer Aktivitätsstörung der Gerinnungsfaktoren VIII (= Hämophilie A) und IX (= Hämophilie B) mit Blutungserscheinungen unterschiedlichen Ausmaßes.
→ Klinisch-relevant: Andere Gerinnungsdefekte, die u.a. die Faktoren II, V, VII, IX und X betreffen, können ein ähnliches klinisches Erscheinungsbild verursachen, werden jedoch nicht zur Hämophilie gezählt.
→ Epidemiologie:
→ I: Die Prävalenz der Hämophilie liegt in Deutschland bei 1/10000 Personen, wobei 80% der Fälle an Hämophilie A und nur 20% an der Hämophilie B leiden.
→ II: Wegen der X-chromosomal-rezessiven Vererbung erkranken Männer (XY) immer, während Frauen (XX) zumeist Konduktorinnen sind und nur sehr selten das charakteristische klinische Bild aufweisen.
→ III: Bei 30% (-50%) der Fälle zeigt sich eine Spontanmutation mit unauffälliger Familienanamnese.
→ Ätiopathogenese: Durch den angeborenen Mangel an Faktor VIII und IX-Aktivität wird das intrinsische plasmatische Gerinnungssystem gestört.
→ I: Hämophilie A: Manifestiert sich infolge eines angeborenen Faktor VIII Mangels. Das Faktor-VIII-Molekül ist ein aus einer leichten - und einer schweren Kette bestehendes Dimer. Es enthält 3A-, 2C- und eine dazwischenliegende inaktive B-Domäne. Die schwere Kette besteht aus 2A und der B-Domäne. Die leichte Kette aus der 3.A- und den zwei C-Domänen.
→ 1) Das Faktor-VIII-Gen liegt auf dem langen Arm des X-Chromosoms und weist eine Vielzahl von Defekten auf, die zu einer Hämophilie A führen können. Insbesondere bei schwerer Hämophilie zeigen sich u.a. große Gendeletionen, Nonsense-Mutationen sowie (relativ häufig) eine Inversion auf dem Intron 22 (Missense-Mutationen an funktionellen Stellen führen zumeist zur leichteren Hämophilie).
→ 2) Faktor VIII wird hauptsächlich in den Hepatozyten synthetisiert.
→ 3) Beim Faktor VIIIa handelt es sich um einen Kofaktor für die Aktivierung von Faktor X mit Hilfe von Faktor IXa in Gegenwart von Kalzium und Phospholipiden. Faktor VIII wiederum wird durch Thrombin aktiviert.
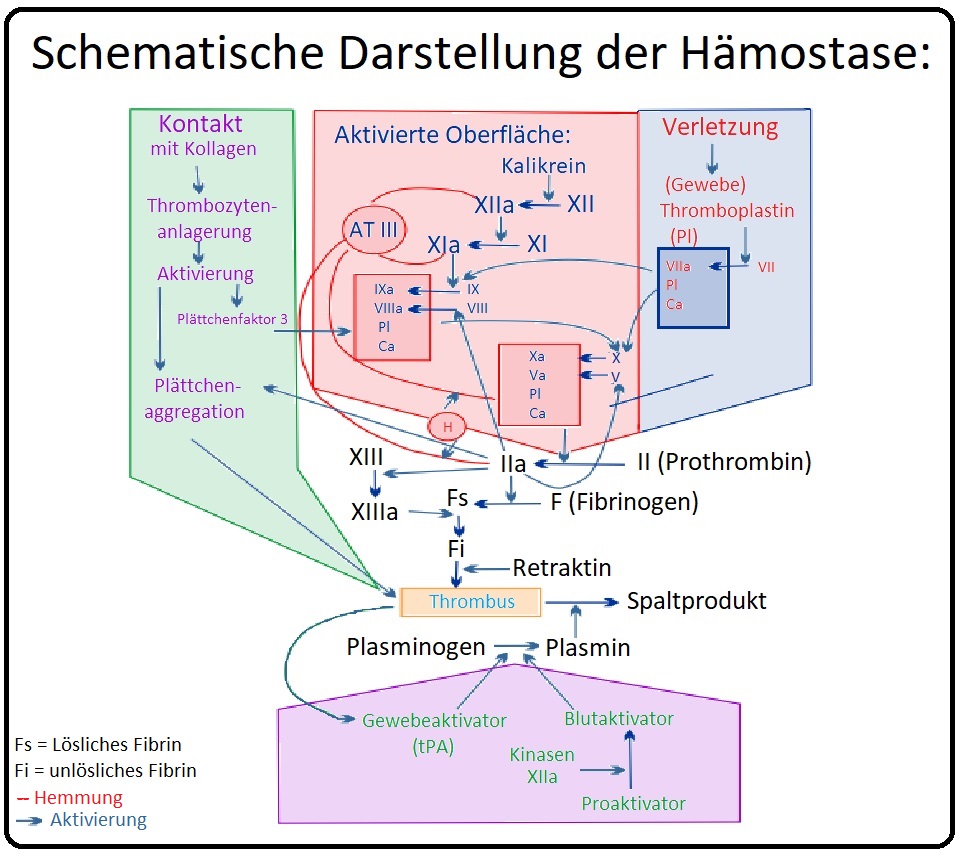
→ II: Hämophilie B: Bei der Hämophilie B besteht ein Mangel an Faktor IX.
→ 1) Beim Faktor IX (Faktor-IX-Gen liegt auch auf dem langen Arm des X-Chromosoms) handelt es sich um ein einkettiges Glykoprotein, das in der Leber Vitamin-K-abhängig synthetisiert wird.
→ 2) Das Molekül besitzt u.a.:

→ 3) Der Faktor IX wird insbesondere durch den Faktor VIIa (weniger des Faktor XIa) in Gegenwart von Kalzium und Phospholipiden aktiviert. Der Faktor IXa wiederum aktiviert in Anwesenheit von Phospholipiden, Kalzium und Faktor VIIIa den Faktor X (X→Xa).
→ III: Pathogenese: Durch den Faktor-Mangel kommt es zu einer verminderten Aktivität des intrinsischen plasmatischen Gerinnungssystems mit konsekutiv reduzierter Aktivierung von Faktor X und Fibrin. Folge ist eine verlangsamte Fibrinbildung und da Thrombin auch den Faktor XIII aktiviert eine gestörte Quervernetzung von Fibrin.
→ Klinik: Das klinische Bild der beiden Hämophilie- Formen ist nicht voneinander zu unterscheiden (vielmehr erfolgt dies durch die laborchemische Bestimmung der Faktoren).
→ I: Erste Hinweise für eine Hämophilie sind ausgedehnte Blutungen nach Bagatelltraumen, spontane Einblutungen in Muskel und Gelenke (Hauptlokalisationen sind Ellenbogen- und Kniegelenk, seltener Schultergelenk), aber auch postoperative Nachblutungen (sowohl Sofortblutungen, als auch besonders charakteristisch Spätblutungen nach Stunden und Tagen).
→ II: Weitere Symptome umfassen schmerzlose Hämaturie (später nach Bildungen von Gerinnseln können sich Koliken entwickeln) und große lokalisierte Hämatome.
→ III: Rezidivierende Gelenk- und Muskelblutungen (insbesondere im Bereich des Unterarm und der Unterschenkelmuskulatur, seltener die Oberschenkelmuskulatur betreffend) führen nicht selten zu sekundären Folgeschädigungen wie u.a.
→ 1) Muskelatrophie und Muskelkontraktur.
→ 2) Chronische Synovitis und Schädigungen des Bandapparates sowie Gelenkversteifung.
→ IV: Subhämophile Patienten mit milder Verlaufsform entwickeln Blutungen nur nach Traumen und größeren Operationen oder Verletzungen.
→ Komplikationen: Wichtige und z.T. schwerwiegende Komplikationen der Hämophilie sind u.a.:
→ I: Massive Einblutungen in die Muskulatur (Kompartmentsyndrom, hypovolämischer Schock) und das Retroperitoneum mit konsekutivem Resorptionsfieber und Leukozytose. (Differenzialdiagnose akutes Abdomen, Appendizitis, etc.)
→ II: Intrakranielle Blutungen sind in bis zu 25% der Fälle Todesursache.
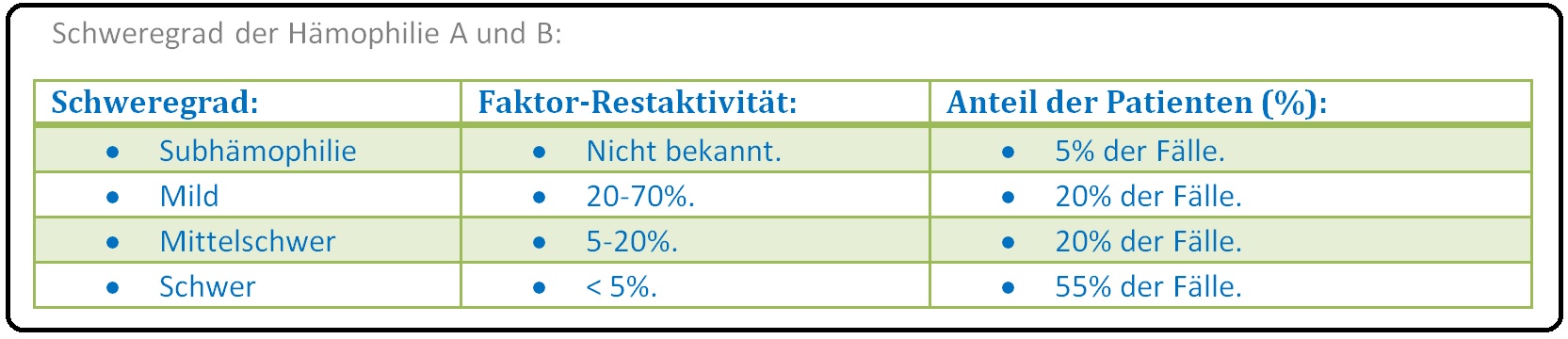
→ Klassifikation: Die Hämophilie A und B lässt sich in 4 klinische Schweregrade unterteilen:
→ Diagnose: Wichtig bei der Diagnose ist die umfangreiche Eigenanamnese mit klinischer Symptomatik (vermehrtes Bluten, Nachblutungen, Gelenkblutungen) sowie die Familienanamnese (evtl. positiv).
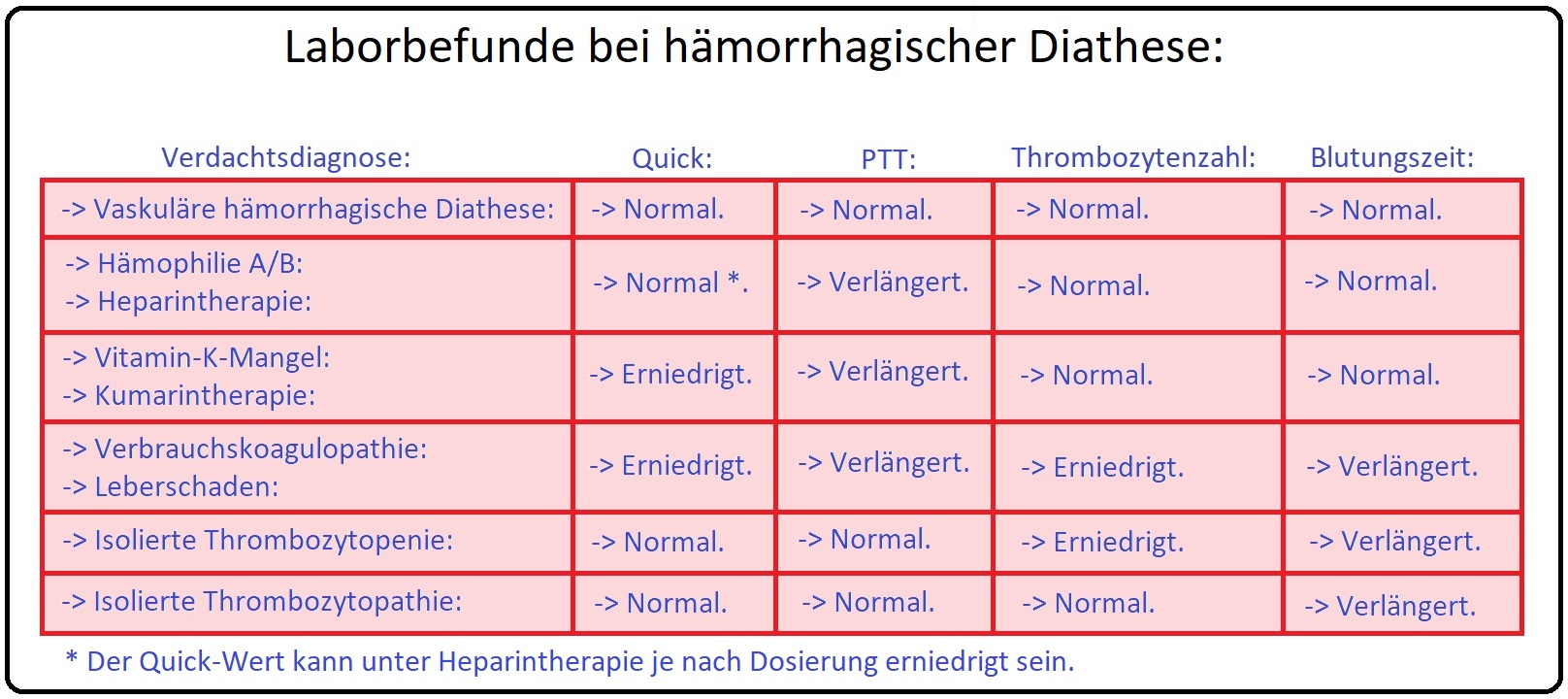
→ I: Laborchemisch ist die verlängerte aPTT richtungsweisend; die primäre Blutstillung bzw. Blutungszeit sowie die Thombin- bzw. Prothombinzeit sind regelrecht (Abb: Wichtige Gerinnungsparameter und ihre Normwerte).

→ II: Zur Sicherung der Diagnosestellung ist die Bestimmung der Faktor VIII- und Faktor IX-Aktivität obligat.
→ III: Schließlich sollte ein kompletter Gerinnungs- und Thrombozytenstatus zur Erfassung der Hämostase-Funktion erfolgen.
→ Differenzialdiagnose: Von der Hämophilie A/B müssen insbesondere nachfolgende Erkrankungen abgegrenzt werden:
→ I: Von-Willebrand-Jürgens-Syndrom.
→ II: Antikörper-bedingter Faktor-VIII-Mangel (= Hemmkörperhämophilie) grenzt sich insbesondere in der Anamnese und dem Nachweis eines Antikörper gegen Faktor VIII ab.
→ Therapie: Die Behandlung der Hämophilie erfolgt primär symptomatisch.
→ I: Allgemeinmaßnahmen: Umfassen u.a. bei äußeren Blutungen Anlage eines Druckverbandes, bei Gelenkblutungen Ruhigstellung, Hochlagerung, Kühlung, etc.
→ II: Prophylaxe: Wichtige Prophylaxen bei Hämophilie-Patienten sind insbesondere:
→ 1) Keine intramuskuläre Injektionen.
→ 2) Keine Applikation von Thrombozytenaggregationshemmern (z.B. ASS), HAES oder Dextranen, etc.
→ III: Substitutionstherapie: Die Applikation von Faktorenkonzentrat ist Mittel der ersten Wahl bei schweren Formen:
→ 1) In der Akuttherapie bei Blutungen ist die sofortige intravenöse Gabe von 25-50IE/kgKG sowie ggf. eine lokale Blutstillung indiziert.
→ Klinisch-relevant: Zur Berechnung der Dosis existiert eine Faustregel; sie besagt, dass eine Einheit Faktor-VIII- oder IX-Konzentrat die Aktivität im Blut um 1% pro kgKG anhebt. Entsprechend berechnet sich die Gesamtmenge, um den Wert z.B. von 50 oder 70% der Norm zu erhalten.

→ 2) Zur Dauertherapie werden bei Hämophilie A 25IE Faktor-VIII-Konzentrat 3x/Woche und bei Hämophilie B 25IE-Faktor IX-Konzentrat 2x/Woche verabreicht.
→ IV: Bei leichteren Formen der Hämophilie A ist bei kleineren Blutungen oder Operationen (z.B. Zahnextraktion) die Gabe von Desmopression (DDAVP) in einer Dosierung von 0,4µg/kgKG über 30min möglich (intravenös alle 12 Stunden über maximal 5 Tage). Bei mittelschwerer Hämophilie im Bedarfsfall z.B. im Rahmen von Blutungskomplikationen steht das Anheben der Gerinnungsfaktoraktivität im Vordergrund.
→ V: Nicht zuletzt ist bei größeren Operationen, größeren Verletzungen, etc. bei allen Formen der Hämophilie eine Faktorensubstitution erforderlich (Zielwert: Faktorenaktivität von 70% der Norm).
→ Klinisch-relevant: Bei der Applikation der Faktor-Konzentraten besteht immer die Gefahr der Entwicklung von unerwünschten Wirkungen; hierzu zählen u.a.:
→ I: Anaphylaxie.
→ II: Infektion mit Hepatitis B (C), HIV (heute nur noch sehr selten).
→ III: Nach häufiger Substitution Ausbildung von Antikörpern mit der Gefahr der Hemmkörperhämophilie.
- Details
- Geschrieben von: CF
- Kategorie: Gerinnungsstörungen
- Zugriffe: 2260
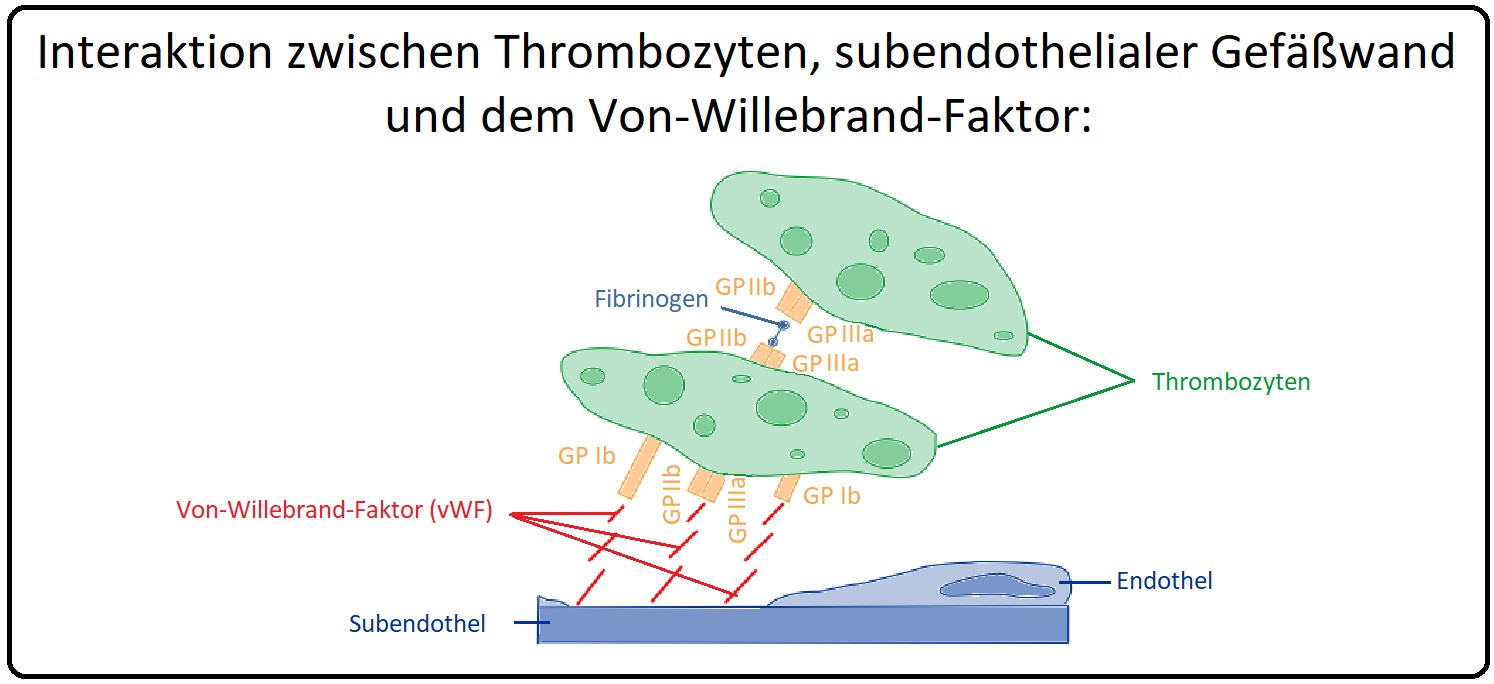
→ Definition: Beim von-Willebrand-Jürgens-Syndrom handelt es sich um eine zumeist autosomal-dominante vererbte Störung des vW-Faktors, die zu einer kombinierten thrombozytären und plasmatischen Gerinnungsstörung führt. Die vermehrte Blutungsneigung entsteht aufgrund des Ausfalls der vWF-Funktion in zweierlei Hinsicht:
→ I: Die Bindung und Stabilisierung des Faktor VIII und
→ II: Die Bindung der Plättchen an das Endothel ist nicht mehr gewährleistet.
→ Epidemiologie: Das Von-Willebrandt-Syndrom stellt die häufigste hereditäre Gerinnungsstörung dar (Gesamtprävalenz 10/100000 Einwohnern), die beide Geschlechter gleichermaßen betreffen kann.
→ I: Die Prävalenz symptomatischer Patienten liegt bei 1,25/10000 Einwohner und
→ II: Die Prävalenz für asymptomatische Fälle liegt bei etwa 1%.
→ Ätiopathogenese:
→ I: Morphologie: Das vWF-Monomer hat ein Molekulargewicht von 309000 Dalton und besitzt genau wie der Faktor VIII drei A-, zwei B- und eine C-Domäne sowie zusätzlich noch vier D-Domänen. Das Gen des vW-Faktors ist auf Chromosom 12 lokalisiert und er wird überwiegend in Endothelzellen und Megakaryozyten synthetisiert. Die Vorstufe, der Pro-Willebrand-Faktor, bildet initial ein Dimer und anschließend Multimere, die in den Weibel-Palade-Bodies (= zytoplasmatische Organellen von Endothelzellen) gespeichert werden. Der von Willebrand-Faktor wird durch Abspaltung des Propeptids freigesetzt. Das vWF-Protein besitzt Bindungsstellen für Faktor VIII, Glykoprotein Ib, IIb und IIIa sowie für Kollagen.
→ II: Beim angeborenen vW-Syndrom können 3 Typen unterschieden werden:
→ 1) Typ I: Stellt mit bis zu 80% der Fälle die häufigste Form dar, wird autosomal-dominant vererbt und ist durch eine quantitative Verminderung des vWF bedingt.
→ 2) Typ II: Diese Form wird ebenfalls autosomal-dominant vererbt und beruht auf einer qualitativen Abnormität des Faktors.
→ 3) Typ III: Wird autosomal-rezessiv vererbt und weist einen deutlich verminderten Spiegel des vWF auf.
→ III: Die Lokalisation des Gendefektes ist insbesondere bei den Typen I und III noch nicht genau bekannt. Beim Typ II wiederum sind einige Punktmutationen bekannt:
→ 1) Subtyp IIa: Es besteht eine Punktmutation in der A2-Domäne. Das betroffene Protein wird durch eine spezifische Plasmaprotease abgebaut (und zirkuliert in verminderter Konzentration).
→ 2) Subtyp IIb: Hierbei zeigt sich eine Punktmutation in der A1-Domäne, die für die GPIb-bindende Domäne kodiert. Folge ist eine ausgeprägte Überaktivität des Proteins mit vermehrter Bindung des mutierten vWF an Plättchen mit konsekutiv vermehrter Elimination (Verlust) des vWF-Plättchen-Komplexes.
→ 3) Subtyp IIn: Hierbei ist eine Mutation am N-terminalen Ende des von-Willebrand-Faktors (= Bindungsstelle des Gerinnungsfaktor VIII). Daraus resultiert eine verminderte Bindungsfähigkeit des vWF für den Faktor VIII, was zum Krankheitsbild der leichten Hämophilie führt.
→ IV: Zudem existiert auch noch ein erworbenes Von-Willebrand-Jürgens-Syndrom; phänotypisch liegt ein Mangel an von-Willebrand-Faktor vor, der für die Funktion des Faktor VIII und Thrombozyten von Bedeutung ist. Ursachen hierfür sind u.a.:
→ 1) Lymphoproliferative Erkrankungen und monoklonale Gammopathie.
→ 2) Maligne Tumoren und Autoimmunerkrankungen.
→ 3) Weitere Ursachen: Sind u.a. Hypothyreose, Medikamente wie Ciprofloxacin und Valproat.
→ Klinisch-relevant: Es existiert eine weitere Klassifikation nach dem Schweregrad des von-Willebrand-Jürgens-Syndrom:

→ Klinik: Das klinische Bild des von-Willebrand-Jürgens-Syndrom ist sehr variabel und reicht von asymptomatischen Verläufen bis hin zum klinischen Bild einer Hämophilie (Schweregrad III).
→ I: Spontane Blutungen wie bei der Hämophilie sind selten, vielmehr manifestieren sich kleiner, diskrete Blutungen wie subkutane Hämatome, Petechien, Schleimhautblutungen, Epitaxis, Neigungen zu Nachblutungen bei Bagatelltraumen, allgemein posttraumatisch und postoperativ (z.B. auch nach Zahnextraktion oder Tonsillektomie, etc.).
→ II: Bei 10% der Betroffenen manifestieren sich Menorrhagien und gastrointestinale Blutungen, insbesondere bei Nachweis von intestinalen Angiodysplasien können diese Blutungen lebensbedrohlich sein.
→ Diagnose: Die Diagnostik des von-Willebrand-Jürgens-Syndroms ist in mehrerer Hinsicht schwierig, da u.a. die Defekte meist leicht sind, es existiert eine große biologische Variabilität bezüglich der Faktor VIII und vW-Faktor-Aktivität, etc. Eine umfangreiche Eigen- und Familienanamnese ist somit obligat.
→ I: Zu den allgemeinen Screeningtests des Von-Willebrand-Jürgens-Syndrom (Abb.: Wichtige Gerinnungsparameter und ihre Normwerte) zählen u.a.:
→ 1) aPTT (= aktivierte partielle Thromboplastinzeit normal oder verlängert).
→ 2) Blutungszeit (verlängert) und
→ 3) PFA (= hierbei handelt es sich um einen Thrombozytenfunktionstest = platelet-function-analysis).
→ Klinisch-relevant: Personen mit der Blutgruppe 0 weisen eine 30%ige niedriger Von-Willebrand-Faktor Aktivität auf als alle anderen.
→ II: Diagnostisch von großer Bedeutung ist die Bestimmung der vWF-Aktivität:
→ 1) Diese wird mit Hilfe des Riskocetin-Cofaktor-Test (Normalwert > 50%) eruiert. Bei Risocetin handelt es sich um ein Antibiotikum, dass in Abhängigkeit von der vWF-Konzentration Thrombozyten aggregiert. Ist die vWF-Aktivität reduziert ist der Test positiv und stellt die Diagnose (eine normale Aktivität schließt das Von-Willebrand-Jürgens-Syndrom weitestgehend aus).
→ 2) Eine Ausnahme bildet die Typ IIn Mutation; hierfür müssen weitere diagnostische Untersuchungen wie z.B. Bestimmung des vWF-Antigens oder Faktor VIII-Aktivität, etc. durchgeführt werden.
→ Differenzialdiagnose: Von dem von-Willebrand-Jügens-Syndrom muss insbesondere die Hämophilie A/B und die Hemmkörperhämophilie abgegrenzt werden. Klinische Unterschiede sind v.a. das Fehlen vom petechialen Blutungstyp sowie keine Verlängerung der Blutungszeit.
→ Therapie:
→ I: Allgemeinmaßnahmen: Bei akuten Blutungen kann ein Druckverband, bei Epitaxis eine Nasentamponade, etc. angelegt werden.
→ II: Spezielle Maßnahmen:
→ 1) Bei Typ I und II (Ausnahme Typ IIb) ist eine Therapie oder Prophylaxe nur bei Operationen, schweren Traumen gastrointestinalen Blutungen indiziert. Die Therapie der ersten Wahl ist die Applikation von Desmopression in einer Dosierung von 0,3-0,4µg/kgKG als Kurzinfusion (alle 12 Stunden über maximal 5 Tage); dies führt zu einem 3-4-fachen Anstieg der Risocetin-Cofaktor- und Faktor-VIII-Aktivität.
→ Klinisch-relevant: Insbesondere bei längerer Applikation besteht die Gefahr der Tachyphylaxie (d.h. zu einem zu geringen oder fehlenden Anstieg von Risocetin-Cofaktor und Faktor-VIII), sodass Prä- und Postinfusionskontrollen obligat sind.
→ 2) Beim Typ IIb und Typ III ist das das Desmopression wirkungslos, sodass eine Indikation für eine Therapie mit Von-Willebrand-Faktor-haltigem Konzentrat indiziert ist. Konzentrate die den vWF enthalten sind Haemate und IMMUNATE, während Faktor-VIII-Konzentrate keinen vWF enthalten und somit nicht für die Therapie geeignet sind.