- Details
- Geschrieben von: CF
- Kategorie: Thrombophilie
- Zugriffe: 2617
→ Definition: Das Antiphospholipid-Syndrom zählt zu den erworbenen Thrombophilien; es handelt sich hierbei um eine Autoimmunerkrankung, die durch das Auftreten von rezidivierenden arteriellen - und venösen Thrombosen, Spontanaborten, Thrombozytopenie mit konsekutivem Nachweis von Antiphospholipid-Antikörpern charakterisiert ist.
→ Epidemiologie: Die Prävalenz für das Antiphospholipid-Syndrom liegt in der Normalbevölkerung bei 2-5%, wobei sie im Alter (durch ein verändertes Immunsystem) deutlich zunimmt.
→ Ätiopathogenese:
→ I: Antiphospholipid-Ak richten sich insbesondere gegen Plasmaproteine, die eine hohe Affinität zu negativ geladenen Oberflächen aufweisen z.B. Phospholipide wie Cardiolipin, Phosphatidylserin, Phosphatidylethanolamin etc; diese sind Bestandteil von Zellmembranen in den verschiedenen Organen (Leber, Herz Gehirn, etc.).
→ II: Wichtige Antikörper hierbei sind:
→ 1) Anti-Beta-Glykoprotein I-Antikörper,
→ 2) Anti-Cardiolipin-Antikörper,
→ 3) Anti-Prothrombin-Antikörper, etc.
→ III: Hauptzielantigene der Antiphospholipid-Antikörper sind u.a.:
→ 1) Apolipoprotein H (= Beta2-GPI) und
→ 2) Das Prothrombin,
→ 3) Weitere Antigene sind zudem aktiviertes Protein C und S, Annexin V, Faktor XII, etc.
→ IV: Pathomechanismus: Die Aktivierung des Gerinnungssystems durch Antiphospholipid-Antikörper ist bis heute nicht genau geklärt, jedoch existieren verschiedene Erklärungsansätze wie:
→ 1) Antiphospholipid-AK beeinträchtigen endogene antikoagulatorische Mechanismen wie z.B. Zerstörung des Annexin-V-Schutzes der Plazenta, Hemmung von Antithrombin oder des Protein-C-Pathways.
→ 2) Es scheint, dass Anti-Phospholipid-AK die Bindung und konsekutive Aktivierung der Thrombozyten (mit Thrombinbildung an der Oberfläche) induzieren.
→ 3) Nicht zuletzt sind Interaktionen mit Endothelzellen möglich. Im Anschluss kommt es zur Expression von Adhäsionsmolekülen und Bindung von Gewebefaktor sowie weiterer Gerinnungsfaktoren.
→ 4) Gerade die Hemmung von TFPI (= Tissue-Factor-Pathway-Inhibitor), der auch an negativen Oberflächen und dem Faktor Xa bindet, führt zu einer vermehrten Thrombin-Produktion.
→ 5) Des Weiteren kommt es zur Hemmung der Fibrinolyse durch Blockade des endothellialen Annorexin-II-Rezeptors, an der tPA (= Tissue-Plasminogen-Activator) und Plaminogen bindet. Das Komplementsystem wird zudem aktiviert.
→ V: Ätiologie: Ursachen, die ein Antiphospholipid-Syndrom induzieren sind u.a.:
→ 1) Autoimmunerkrankungen wie systemischer LE, rheumatoide Arthritis, Sjögren-Syndrom, Immunthrombozytopenie.
→ 2) Antiphospholipid-Antikörper können sich auch im Rahmen von Infektionen wie Lyme-Borreliose, Varizella-Zoster-Virus-, Hepatitis C- und HIV-Infektionen, etc.
→ 3) Medikamenteninduziert: Durch Antibiotika (Penicilline und Derivate sowie Streptomycin), Antihypertensiva (z.B. Beta-Blocker insbesondere Propranolol, Hydralazin), Antiarrhythmika (Chinidin, Procainamid) und nicht zuletzt Antikovulsiva bzw. Psychopharmaka wie Valproat und Chlorpromazin (Neuroleptika).
→ 4) Nicht zuletzt existiert das sehr seltene familiäre Antiphospholipid-Syndrom, das einen autosomal-dominanten Erbgang aufweist.
→ V: Klassifikation: Bei dem Antiphospholipid-Syndrom kann zudem hinsichtlich der Ätiopathogenese 2 Formen unterschieden werden:
→ 1) Primäres Antiphospholipid-Syndrom mit idiopathischer Genese und einer
→ 2) Sekundären Form im Rahmen einer Grunderkrankung wie z.B. Kollagenosen wie v.a. systmischer Lupus erythematodes, aber auch Tumoren, etc.
→ Klinik: Das Antiphospholipid-Syndrom ist eine Multiorganerkrankung, die mit verschiedenen Symptomen assoziiert ist:
→ I: Thrombosen kleiner und größerer Gefäße in den verschiedenen Organe, aber auch atypische Lokalisationen. Es können sowohl venöse als auch arterielle Thrombosen nachgewiesen werden. Klinische Korrelate sind u.a.:
→ 1) Tiefe Beinvenenthrombose und Lungenembolie, aber auch Myokardinfarkt.
→ 2) Neurologische Symptome mit Migräne, Seh- evtl. Hörverlust bis hin zur TIA.
→ 3) Dermatologische Auffälligkeiten wie Livedo reticularis und Hautulzerationen, Raynaud-Syndrom, etc.
→ II: Bei schwangeren Frauen mit dem APS kommt es nicht selten durch Mikrothromben in der Plazenta zu Aborten; bei 30-40% der Frauen manifestieren sich sogar rezidivierende Aborte.
→ III: Eine sehr selten bestehende Thrombopenie kann eine Blutungsneigung induzieren.
→ IV: Komplikation: Das „catastrophic“ Antiphospholipid-Syndrom ist eine durch Mikrothromben induzierte lebensbedrohliche Situation, die zu einem Multiorganversagen führen kann. Triggermechanismen hierfür sind u.a. Operationen, Trauma, Infektionen, Phase des Absetzens von Vitamin-K-Antagonisten (Marcumar). Differenzialdiagnostisch müssen eine schwere Sepsis, disseminierte intravasale Koagulopathie und nicht zuletzt die Heparin-induzierte Thrombozytopenie ausgeschlossen werden.
→ Diagnose:
→ I: Bei Vorliegen einer Thrombose, unklaren Aborten, arteriellen Thrombosen ohne arteriosklerotische Veränderungen, unklaren Thrombozytopenien und nicht zuletzt Autoimmunerkrankungen mit Gefäßbeiteiligung sollte immer auch eine Untersuchung nach Antiphospholipid-Antikörper erfolgen.

→ Klinisch-relevant: Unter dem Oberbegriff der Antiphospholipid-Antikörper werden Lupusantikoagulans (LA) Antikardiolipin, Antikörper (aCL) und Anti-ß2-GPI-Antikörper sowie weitere AK (siehe Tabelle) zusammengefasst.
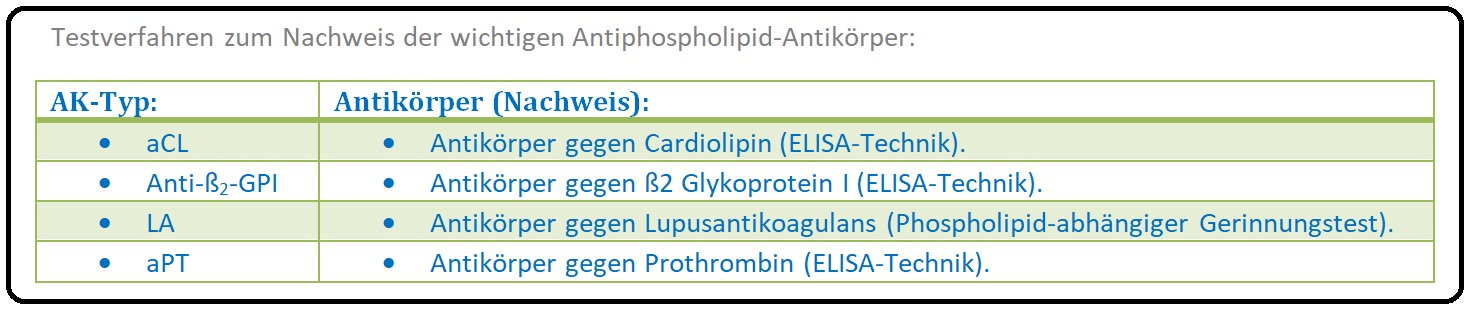
→ II: Labor: Antiphospholipid-Antikörper sind ein Gemisch aus Autoantikörpern vom IgM und/oder IgG, seltener vom IgA-Typ. Diese AK sind Inhibitoren des Gerinnungssystems und sind gegen Phospholipid-Protein-Komplexe gerichtet (konkreter gegen Plasmaproteine, die an anionische Phospholipid-Oberflächen binden). Zu diesen sogenannten „Target-Proteinen“ zälhen u.a. das ß2-Glykoprotein und das Prothrombin. Es stehen 2 unterschiedliche Testsysteme für die Diagnostik der Antiphospholipid-AK zur Verfügung:
→ 1) Nachweis der Verlängerung von Gerinnungszeiten Phospholipid-abhängiger Gerinnungstests und anschließender Normalisierung bei Zugabe von Phospholipiden im Überschluss. Dieses Verfahren wird als sogenannter „Lupusantikoagulans-Test“ bezeichnet.
→ 2) ELISA: ELISA-Systeme dienen dem Erkennen insbesondere der Anticardiolipin und Anti-ß2-GPI-Antikörpern.
→ Klinisch-relevant:
→ A) Die Diagnose der Antiphospholipid-Antikörper wird laborchemisch gestellt durch einen positiven Test und die Bestätigung nach 12 Wochen.
→ B) LA-Antikörper fallen häufig durch eine verlängerte aPTT auf, jedoch schließt eine normale aPTT ein Lupusantikoagulans nicht aus.
→ Therapie:
→ I: Bei asymptomatischen Patienten mit wiederholten hochtitrigem Phospholipid-Antikörpern-Nachweis wird eine prophylaktische Therapie ASS-Dauertherapie empfohlen.
→ II: In Situationen mit erhöhtem Risiko ist bei Patienten mit Antiphospholipid-Syndrom eine bedarfsangepasste Thromboseprophylaxe mit Kompressionsstrümpfen und Heparin obligat.
→ III: In der akuten Phase der thromboembolischen Erkrankung eine adäquate Thrombosetherapie mit anschließender oraler Antikoagulation (Marcumar) mit einem INR von 2-3 indiziert. Da das Rezidivrisiko bei Patienten mit APS um ein vielfaches höher ist als z.B. beim Faktor-V-Leiden- oder Prothrombin-Genmutation, wird heutzutage eine langfristige orale antikoagulatorische Behandlung empfohlen.
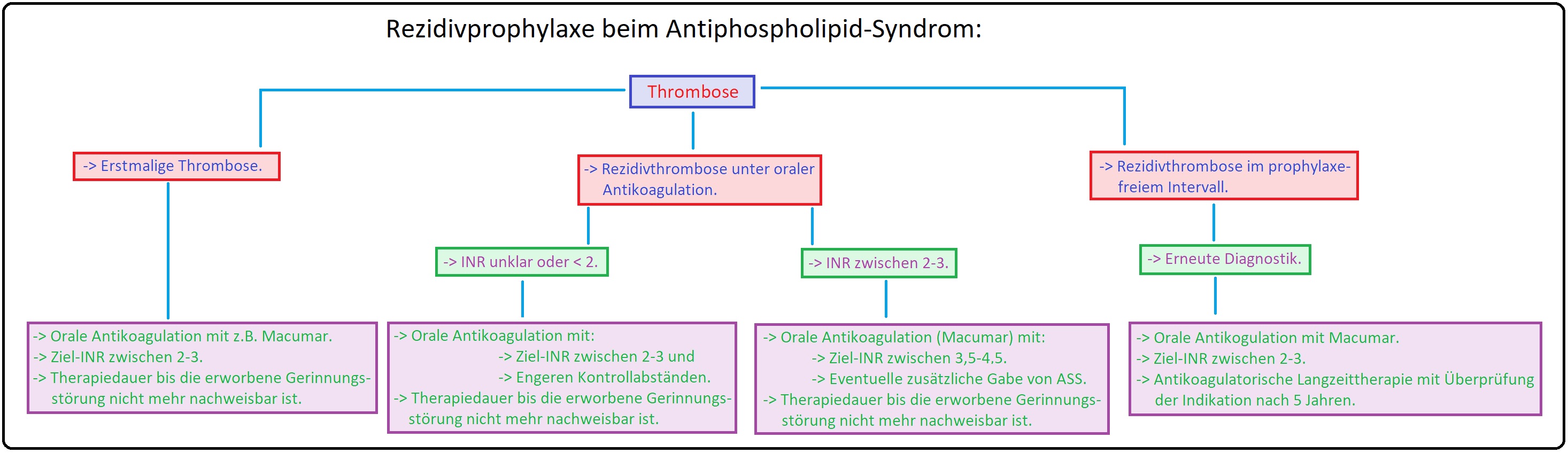
→ IV: Bei habituellen Aborten (APS assoziiert) wird eine langfristige Kombinationstherapie aus ASS und niedermolekularem Heparin in niedriger Dosierung appliziert.
→ V: Catastrophic APS: Liegt diese schwere Verlaufsform vor und sind mehr als 3 Organsysteme betroffen werden Plasmapherese, hochdosierte Glukokortikoide, hochdosierte (intravenöse) Immunglobuline sowie Cyclophosphamid eingesetzt.
- Details
- Geschrieben von: CF
- Kategorie: Thrombophilie
- Zugriffe: 2254
→ Definition: Bei der Thombophilie handelt es sich um eine hereditäre oder erworbene Gerinnungsstörung, bei der es zum vermehrten Auftreten von thromboembolischen Ereignissen kommt. Abhängig vom Auftreten einer charakteristischen klinischen Symptomatik differenziert man zwischen einer asymptomatischen und symptomatischen (= klinisch manifesten) Form.
→ Ätiopathogenese: Physiologische Inhibitoren der Hämostase begrenzen und kontrollieren die Gerinnungsaktivität und verhindert somit einer überschießende Fibrin-Bildung in den Gefäßen. Manifestieren sich diesbezüglich Verminderungen bzw. Defekte der Inhibitoren so kommt es zur Hyperkoagulabilität (die überwiegend das venöse System betreffen). Nach dem Entstehungsmechanismus unterscheidet man zwischen einer angeborenen und erworbenen Thrombophilie.
→ Klinik: Zu den charakteristischen Symptomen einer Thrombophilie zählen insbesondere:
→ I: Frühes Alter der Erstmanifestation (zumeist vor dem 40. Lebensjahr).
→ II: Spontanthrombosen oder im Verhältnis zum Stimulus mit überschießender Ausprägung.
→ III: Ungewöhnliche, wechselnde oder ausgedehnte Lokalisationen.
→ IV: Weitere Symptome: Sind u.a.:
→ 1) Häufige Rezidive,
→ 2) Eine deutliche familiäre Disposition.
→ 3) Abortneigung, etc.
→ Klassifikation: Bei der Thrombophilie wird abhängig von der Pathogenese zwischen 2 Formen unterschieden:
→ I: Angeborene Thrombophilie: Hierunter wird eine genetisch-determinierte Neigung zur Thromboembolien verstanden. Die klinische Manifestation ist schließlich multifaktoriell bedingt und wird durch das Hinzukommen von weiteren Risikofaktoren getriggert. Ursachen der angeborenen Form sind insbesondere:
→ 1) Mangel an natürlichen antikoagulatorischen Protein wie. z.B. Antithrombin-, Protein-C- und Protein-S-Mangel.
→ 2) Mit vermehrter Häufigkeit Mutationen von Gerinnungsfaktoren wie z.B. Faktor-V-Leiden-, Prothrombin-, Genmutation und
→ 3) Weitere hereditäre Mechanismen wie die kongenitale Dysfibrinogenämie.

→ II: Erworbene Thrombophilie: Verschiedene klinische Entitäten gehen mit einem erhöhten Risiko für die Entwicklung von thromboembolischen Ereignissen (venös, aber auch arteriell) einher. Hierzu zählen insbesondere:
→ 1) Antiphospholipid-Syndrom,
→ 2) Heparin-induzierte Thrombozytopenie,
→ 3) Schwangerschaftsassoziiertes erhöhtes Thromboserisiko.
→ 4) Nephrotisches Syndrom: Beim nephrotischen Syndrom zeigt sich einer erhöhte Thromboseneigung, deren Ursache jedoch noch nicht genau bekannt sind. Angenommen werden u.a. Verminderung der Gerinnungsinhibitoren (wie Antithrombin, Protein-C und -S), Zunahme der Blutviskosität und der Thrombozytenzahl sowie Verminderung der fibrinolytischen Kapazität. Klinisches Korrelat des thrombotischen Ereignisses ist die Nierenvenenthrombose, die sich insbesondere beim nephrotischen Syndrom im Rahmen einer Glomerulonephritis manifestiert, aber Thrombosen anderer Lokalisation sind möglich. Das Thromboserisiko ist besonders hoch bei Serumalbumin < 20g/l, Fibrinogen > 4,0 und das Antithrombin < 50g/l.

→ Diagnose: Im Vordergrund der Diagnosestellung steht die laborchemische Untersuchung (die Laboranalytik bestätigt in 60% der Fälle die Diagnose; eine negative Laboranalytik schließt die Diagnose der Thrombophilie nicht aus).
→ I: Anamnese/klinische Untersuchung: Hierbei stehen Eigenanamnese (Medikamenteneinnahme, junges Alter bei Erstmanifestation, vorherige bzw. rezidiverende Thrombose wie tiefe Beinvenenthrombose, atypische Thromboselokalisationen wie Budd-Chiari-Syndrom, Sinusvenenthrombose, Pfortaderthrombose, etc.) und die Familienanamnese im Vordergrund.

→ II: Labor:
→ 1) Nach folgende Laborparameter sind mit einem thromboembolischen Ereignis assoziiert:

→ 2) Als Basisdiagnostik gilt für die Thrombophelie Bestimmung von Faktor-V-Leiden-Mutation, APC-Resistenz, Prothrombinmutation, Antithrombin-III, Lupus-Antikoagulans, etc.
→ 3) Die erweiterte Diagnostik umfasst dann die Bestimmung von Faktor VIII, Protein-C und S sowie Homocystein.
→ Therapie: Liegt eine manifeste Thrombophilie vor, ist das therapeutische Ziel ein Rezidiv zu vermeiden. Mittel der Wahl ist die orale Antikoagulation meist mittels Macumar mit einem Ziel-INR zwischen 2-3. Die Therapiedauer richtet sich insbesondere nach dem Thromboserisiko. Wird die orale Antikoagulation beendet ist eine risikoadaptierte Thromboseprophylaxe mit einem niedermolekularen Heparin obligat.
- Details
- Geschrieben von: CF
- Kategorie: Thrombophilie
- Zugriffe: 3823
→ Definition: Bei der Faktor-V-Leiden-Mutation handelt es sich um eine hereditäre Thrombophilie mit einer Punktmutation im Codon 1691 (Exon 10) des Faktor-V-Gens mit konsektiv erhöhter Thromboseneigung; diese Mutation stellt definitiongemäß einen Polymorphismus dar.
→ Epidemiologie:
→ I: Die Faktor-V-Leiden-Genmutation stellt die häufigste genetische Ursache für venöse Thromboembolien (v.a. tiefe Beinvenenthrombose) dar.
→ II: Die Faktor-V-Leiden-Mutation tritt in Europa in 5%-10% der Fälle auf (auch in Deutschland besteht eine erhöhte Disposition), wobei sich ein deutliches Nord-Süd-Gefälle herauskristallisiert (mit deutlich höherer Prävalenz in den nördlichen Regionen und sehr geringe Prävalenz in Regionen wie Asien und Afrika).
→ III: Die Vererbung erfolgt co-dominant, sodass das Thromboserisiko bei homozygoten deutlich höher ist als bei heterozygoten Trägern.
→ 1) Heterozygote Träger haben eine 5-10-fach erhöhtes Thromboserisiko und
→ 2) Homozygote Träger weisen ein 50-100-fach erhöhtes Thromboserisiko auf.
→ IV: Etwa die Hälfte aller Patienten mit familiärer Thromboseneigung sind ist Träger dieser Mutation.
→ Klinisch-relevant: Zudem weisen Menschen mit Faktor-V-Leiden ein 2-4-fach erhöhtes Risiko für Fehlgeburten auf. Die Einnahme von Kontrazeptiva erhöht das Thromboserisiko bei heterozygoten um das 30-fache, bei homozygoten Trägern über das 200-fache.
→ Physiologie: Beim Faktor V des Gerinnungssystems handelt es sich um ein Glykoprotein mit einem Molekulargewicht von 330000 Da, der eine inaktive Vorstufe und einen aktiven Faktor Va aufweist, der als Kofaktor der Serinprotease Faktor Xa fungiert. Der Faktor Va stell eine sogenanntes Akzelerationsglobulin dar, durch deren Präsenz die enzymatischen Prozesse massiv beschleunigt werden (die gleiche Funktion weist der Faktor VIIIa auf).
→ I: Der Faktor V ist der bekannteste prokoagulatorische Faktor und wird durch Thrombin zu Faktor Va aktiviert. Erst der Faktor Va erweist sich als koagulatorisch, indem es die Aktivierung von Prothrombin zu Thrombin um das 1000-fache beschleunigt. Zudem bildet es zusammen mit Faktor Xa den sogenannten "Prothrombinase-Komplex" (+ Ca2+-Ionen) auf der Phospholipidoberfläche von Thrombozyten.
→ II: Aktiviertes Protein C: (= APC)
→ 1) Frühzeitig erfolgt der proteolytische Abbau des Faktor Va durch das aktivierte Protein C mit Hilfe seines Kofaktors Protein S, sodass er seine Akzelerationsfunktion verliert.
→ 2) Des Weiteren spaltet das APC den auf den Phospholipidoberflächen befindliche nicht-aktivierte Faktor V zu Faktor Vac (ac = anticoagulant), der die prokoagulatorischen Eigenschaften verliert und zusammen mit Protein S zu einem Kofaktor des aktivierten Protein C wird. Die Funktion des gebildeten Faktor Vac ist insbesondere ist die Inaktivierung von Faktor VIIIa.
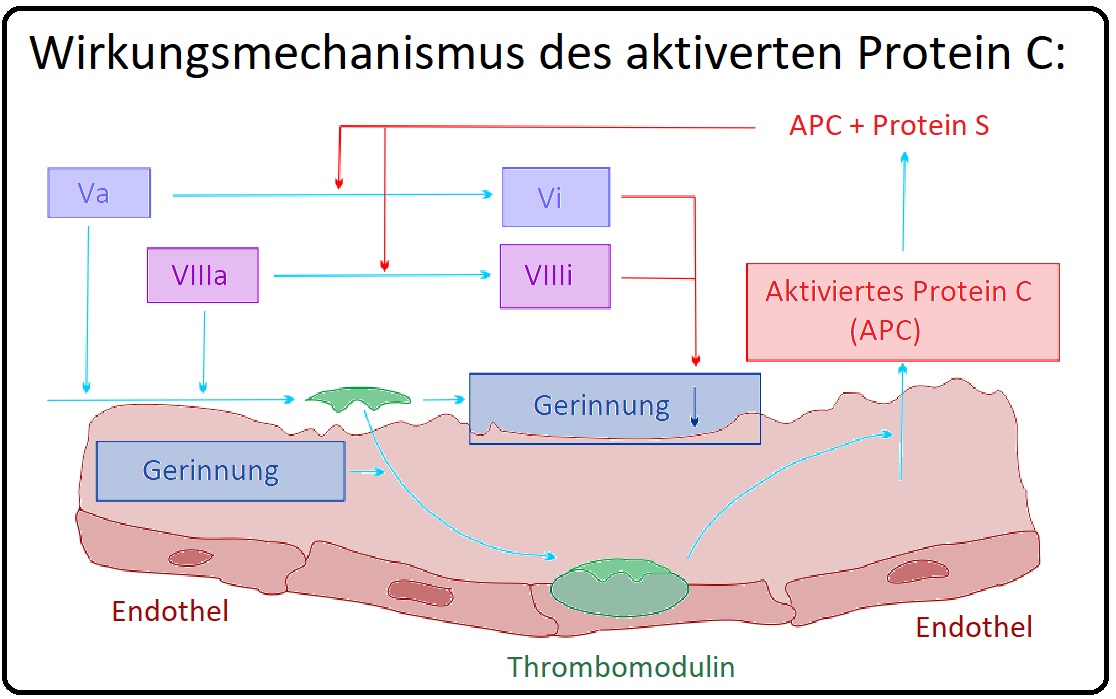
→ III: Das Faktor-V-Gen befindet sich auf Chromosom 1q21-25 und wird überwiegend in der Leber von Hepatozyten gebildet. In Thrombozyten werden 20% des zirkulierenden Faktor V in leicht veränderter Form, als Multimerin I, in der Alpha-Granula gespeichert (Megakaryozyten synthetisieren auch einen geringen Teil des Faktor V).
→ Pathophysiologie: Die Hyperkoagulabilität des Faktor-V-Leiden hat insbesondere 2 Pathomechanismen:
→ I: Durch die Punktmutation kommt es in Position 506 zu einem Austausch von Arginin durch Glutamin. Hierdurch erkennt das aktivierte Protein C (= APC) die Spaltstelle im Faktor Va nicht mehr (aufgehobene Metabolisierung), sodass der Faktor Va seine Aktivität behält und vermehrt Thrombin gebildet wird. Patienten mit dieser Mutation weisen eine deutlich verlangsamte Inaktivierung des aktivierten Faktor V durch das aktivierte Protein C auf. (= APC-Resistenz).
→ II: Intakter Faktor-V dient als Cofaktor für die Inaktivierung der Faktoren VIIIa und Xa. Patienten mit der Faktor-V-Leiden-Genmutation weisen somit auch eine verminderte antikoagulatorische Aktivität des aktivierten Protein C gegenüber Faktor VIIIa und Xa.
→ Klinik:
→ I: Deutlich erhöhte Thromboseneigung (wie tiefe Beinvenenthrombose) insbesondere bei jüngeren Patienten, wenn vor allem zusätzlich erworbene Risikofaktoren wie z.B. Einnahme von Ovulationshemmern, Schwangerschaft, Immobilisation, Trauma, etc. hinzukommen.
→ II: Komplikationen: Wie u.a.:
→ 1) Lungenembolie,
→ 2) Entwicklung einer Sinusvenenthrombose mit diffus drückende Kopfschmerzen, Schwäche im Bereich der Arme bis hin zu Grand-mal-Anfällen, etc.
→ Diagnose.: Dient dem Nachweis einer Thrombophilie und umfasst insbesondere:
→ I: Anamnese und klinische Untersuchung mit Eigen- und Familienanamnese (u.a. Medikamentenanamnese, Vorerkrankungen, Thromboembolien in der Eigenanamnese bzw. Familien-, etc.).
→ II: Labor:
→ 1) APCR-Funktionstest: Stellt eine Variante der aPTT dar und besteht aus einer 2-fachen aPTT-Bestimmung ohne und mit Zusatz einer standardisierten Menge von aktivierten Protein C (APC-Ratio). Bei Vorliegen einer APC-Resistenz beträgt die Verlängerung der aPTT nach Zusatz des aktivierten Protein C weniger als das 2-fache gegenüber dem Ausgangswert.

→ 2) Genotypisierung: Wurde eine APC-Resistenz festgestellt sollte anschließend eine Genotypisierung mittels PCR (= Polymerase-Ketten-Reaktion) erfolgen, um zu eruieren, ob eine homo- oder heterozygote Faktor-V-Mutation vorliegt.
→ Therapie:
→ I: Eine medikamentöse Akuttherapie ist immer im Rahmen einer bestehenden Thrombose oder Embolie indiziert (siehe u.a. tiefen Beinvenenthrombose, Lungenembolie, Sinusvenenthrombose, etc.).
→ II: Ansonsten stehen insbesondere Primär- und Sekundärprophylaxe im Vordergrund:
→ 1) Primärprophylaxe: Hierbei sollte z.B. schwangeren Frauen mit zusätzlichen Risikosituationen z.B. Immobilisation niedermolekulares Heparin prophylaktisch appliziert werden. Auch eine postmenopausale Hormonsubstitution erhöht das Thromboserisiko bei Faktor-V-Genmutation deutlich, sodass die Hormone nicht oral, sondern vielmehr transdermal verabreicht werden sollten, da bei dieser Applikationsform (nach neueren Studien zufolge) wohl kein Thromboserisiko bestehe.
→ 2) Sekundärprophylaxe: Die Rezidivrate von Thromboembolien im Rahmen einer Faktor-V-Genmutation ist erhöht, sodass eine orale Antikoagulation wie bei anderen Patienten über einen Zeitraum von 6-12 Monaten indiziert ist. Jedoch kann eine idiopathische Venenthrombose eine Langzeittherapie mit Macumar induzieren. Hierbei ist insbesondere die Bestimmung der D-Dimere von Bedeutung (ein negativer D-Dimer-Wert 4 Wochen nach Abschluss der oralen Antikoagulation spricht für eine nur sehr geringe Rezidiv-Wahrscheinlichkeit). Vor allem bei rezidivierenden Thrombosen nach längerfristiger Antikoagulation ist eine risikoadaptierte Prophylaxe zu empfehlen (z.B. bei Immobilisation bei z.B. 4-stündigen Flügen sind neben Kompressionsstrümpfen noch niedermolekulare Heparine obligat).

→ 3) Ein zusätzlicher thrombophiler Defekt erhöht das Thromboserisiko wiederum gravierend, sodass zumeist eine prolongierte Antikoagulation indiziert ist.