→ Definition: Das Antiphospholipid-Syndrom zählt zu den erworbenen Thrombophilien; es handelt sich hierbei um eine Autoimmunerkrankung, die durch das Auftreten von rezidivierenden arteriellen - und venösen Thrombosen, Spontanaborten, Thrombozytopenie mit konsekutivem Nachweis von Antiphospholipid-Antikörpern charakterisiert ist.
→ Epidemiologie: Die Prävalenz für das Antiphospholipid-Syndrom liegt in der Normalbevölkerung bei 2-5%, wobei sie im Alter (durch ein verändertes Immunsystem) deutlich zunimmt.
→ Ätiopathogenese:
→ I: Antiphospholipid-Ak richten sich insbesondere gegen Plasmaproteine, die eine hohe Affinität zu negativ geladenen Oberflächen aufweisen z.B. Phospholipide wie Cardiolipin, Phosphatidylserin, Phosphatidylethanolamin etc; diese sind Bestandteil von Zellmembranen in den verschiedenen Organen (Leber, Herz Gehirn, etc.).
→ II: Wichtige Antikörper hierbei sind:
→ 1) Anti-Beta-Glykoprotein I-Antikörper,
→ 2) Anti-Cardiolipin-Antikörper,
→ 3) Anti-Prothrombin-Antikörper, etc.
→ III: Hauptzielantigene der Antiphospholipid-Antikörper sind u.a.:
→ 1) Apolipoprotein H (= Beta2-GPI) und
→ 2) Das Prothrombin,
→ 3) Weitere Antigene sind zudem aktiviertes Protein C und S, Annexin V, Faktor XII, etc.
→ IV: Pathomechanismus: Die Aktivierung des Gerinnungssystems durch Antiphospholipid-Antikörper ist bis heute nicht genau geklärt, jedoch existieren verschiedene Erklärungsansätze wie:
→ 1) Antiphospholipid-AK beeinträchtigen endogene antikoagulatorische Mechanismen wie z.B. Zerstörung des Annexin-V-Schutzes der Plazenta, Hemmung von Antithrombin oder des Protein-C-Pathways.
→ 2) Es scheint, dass Anti-Phospholipid-AK die Bindung und konsekutive Aktivierung der Thrombozyten (mit Thrombinbildung an der Oberfläche) induzieren.
→ 3) Nicht zuletzt sind Interaktionen mit Endothelzellen möglich. Im Anschluss kommt es zur Expression von Adhäsionsmolekülen und Bindung von Gewebefaktor sowie weiterer Gerinnungsfaktoren.
→ 4) Gerade die Hemmung von TFPI (= Tissue-Factor-Pathway-Inhibitor), der auch an negativen Oberflächen und dem Faktor Xa bindet, führt zu einer vermehrten Thrombin-Produktion.
→ 5) Des Weiteren kommt es zur Hemmung der Fibrinolyse durch Blockade des endothellialen Annorexin-II-Rezeptors, an der tPA (= Tissue-Plasminogen-Activator) und Plaminogen bindet. Das Komplementsystem wird zudem aktiviert.
→ V: Ätiologie: Ursachen, die ein Antiphospholipid-Syndrom induzieren sind u.a.:
→ 1) Autoimmunerkrankungen wie systemischer LE, rheumatoide Arthritis, Sjögren-Syndrom, Immunthrombozytopenie.
→ 2) Antiphospholipid-Antikörper können sich auch im Rahmen von Infektionen wie Lyme-Borreliose, Varizella-Zoster-Virus-, Hepatitis C- und HIV-Infektionen, etc.
→ 3) Medikamenteninduziert: Durch Antibiotika (Penicilline und Derivate sowie Streptomycin), Antihypertensiva (z.B. Beta-Blocker insbesondere Propranolol, Hydralazin), Antiarrhythmika (Chinidin, Procainamid) und nicht zuletzt Antikovulsiva bzw. Psychopharmaka wie Valproat und Chlorpromazin (Neuroleptika).
→ 4) Nicht zuletzt existiert das sehr seltene familiäre Antiphospholipid-Syndrom, das einen autosomal-dominanten Erbgang aufweist.
→ V: Klassifikation: Bei dem Antiphospholipid-Syndrom kann zudem hinsichtlich der Ätiopathogenese 2 Formen unterschieden werden:
→ 1) Primäres Antiphospholipid-Syndrom mit idiopathischer Genese und einer
→ 2) Sekundären Form im Rahmen einer Grunderkrankung wie z.B. Kollagenosen wie v.a. systmischer Lupus erythematodes, aber auch Tumoren, etc.
→ Klinik: Das Antiphospholipid-Syndrom ist eine Multiorganerkrankung, die mit verschiedenen Symptomen assoziiert ist:
→ I: Thrombosen kleiner und größerer Gefäße in den verschiedenen Organe, aber auch atypische Lokalisationen. Es können sowohl venöse als auch arterielle Thrombosen nachgewiesen werden. Klinische Korrelate sind u.a.:
→ 1) Tiefe Beinvenenthrombose und Lungenembolie, aber auch Myokardinfarkt.
→ 2) Neurologische Symptome mit Migräne, Seh- evtl. Hörverlust bis hin zur TIA.
→ 3) Dermatologische Auffälligkeiten wie Livedo reticularis und Hautulzerationen, Raynaud-Syndrom, etc.
→ II: Bei schwangeren Frauen mit dem APS kommt es nicht selten durch Mikrothromben in der Plazenta zu Aborten; bei 30-40% der Frauen manifestieren sich sogar rezidivierende Aborte.
→ III: Eine sehr selten bestehende Thrombopenie kann eine Blutungsneigung induzieren.
→ IV: Komplikation: Das „catastrophic“ Antiphospholipid-Syndrom ist eine durch Mikrothromben induzierte lebensbedrohliche Situation, die zu einem Multiorganversagen führen kann. Triggermechanismen hierfür sind u.a. Operationen, Trauma, Infektionen, Phase des Absetzens von Vitamin-K-Antagonisten (Marcumar). Differenzialdiagnostisch müssen eine schwere Sepsis, disseminierte intravasale Koagulopathie und nicht zuletzt die Heparin-induzierte Thrombozytopenie ausgeschlossen werden.
→ Diagnose:
→ I: Bei Vorliegen einer Thrombose, unklaren Aborten, arteriellen Thrombosen ohne arteriosklerotische Veränderungen, unklaren Thrombozytopenien und nicht zuletzt Autoimmunerkrankungen mit Gefäßbeiteiligung sollte immer auch eine Untersuchung nach Antiphospholipid-Antikörper erfolgen.

→ Klinisch-relevant: Unter dem Oberbegriff der Antiphospholipid-Antikörper werden Lupusantikoagulans (LA) Antikardiolipin, Antikörper (aCL) und Anti-ß2-GPI-Antikörper sowie weitere AK (siehe Tabelle) zusammengefasst.
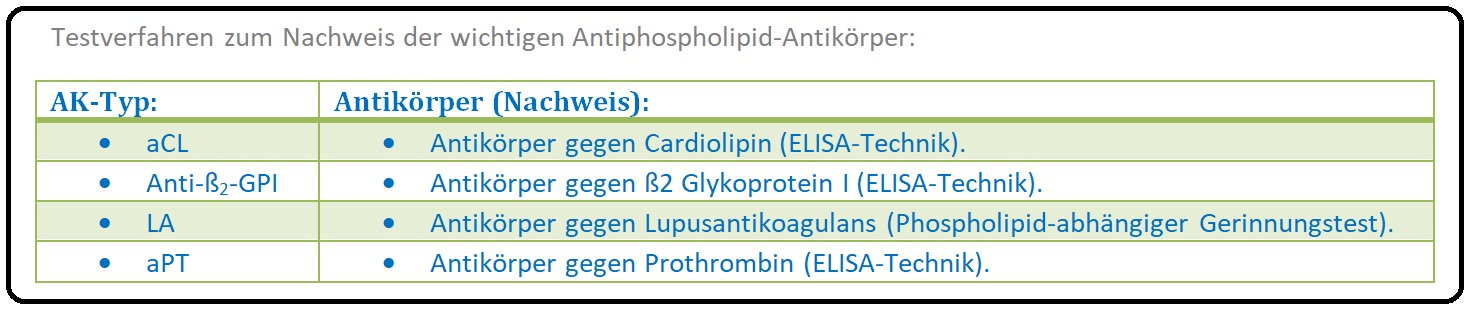
→ II: Labor: Antiphospholipid-Antikörper sind ein Gemisch aus Autoantikörpern vom IgM und/oder IgG, seltener vom IgA-Typ. Diese AK sind Inhibitoren des Gerinnungssystems und sind gegen Phospholipid-Protein-Komplexe gerichtet (konkreter gegen Plasmaproteine, die an anionische Phospholipid-Oberflächen binden). Zu diesen sogenannten „Target-Proteinen“ zälhen u.a. das ß2-Glykoprotein und das Prothrombin. Es stehen 2 unterschiedliche Testsysteme für die Diagnostik der Antiphospholipid-AK zur Verfügung:
→ 1) Nachweis der Verlängerung von Gerinnungszeiten Phospholipid-abhängiger Gerinnungstests und anschließender Normalisierung bei Zugabe von Phospholipiden im Überschluss. Dieses Verfahren wird als sogenannter „Lupusantikoagulans-Test“ bezeichnet.
→ 2) ELISA: ELISA-Systeme dienen dem Erkennen insbesondere der Anticardiolipin und Anti-ß2-GPI-Antikörpern.
→ Klinisch-relevant:
→ A) Die Diagnose der Antiphospholipid-Antikörper wird laborchemisch gestellt durch einen positiven Test und die Bestätigung nach 12 Wochen.
→ B) LA-Antikörper fallen häufig durch eine verlängerte aPTT auf, jedoch schließt eine normale aPTT ein Lupusantikoagulans nicht aus.
→ Therapie:
→ I: Bei asymptomatischen Patienten mit wiederholten hochtitrigem Phospholipid-Antikörpern-Nachweis wird eine prophylaktische Therapie ASS-Dauertherapie empfohlen.
→ II: In Situationen mit erhöhtem Risiko ist bei Patienten mit Antiphospholipid-Syndrom eine bedarfsangepasste Thromboseprophylaxe mit Kompressionsstrümpfen und Heparin obligat.
→ III: In der akuten Phase der thromboembolischen Erkrankung eine adäquate Thrombosetherapie mit anschließender oraler Antikoagulation (Marcumar) mit einem INR von 2-3 indiziert. Da das Rezidivrisiko bei Patienten mit APS um ein vielfaches höher ist als z.B. beim Faktor-V-Leiden- oder Prothrombin-Genmutation, wird heutzutage eine langfristige orale antikoagulatorische Behandlung empfohlen.
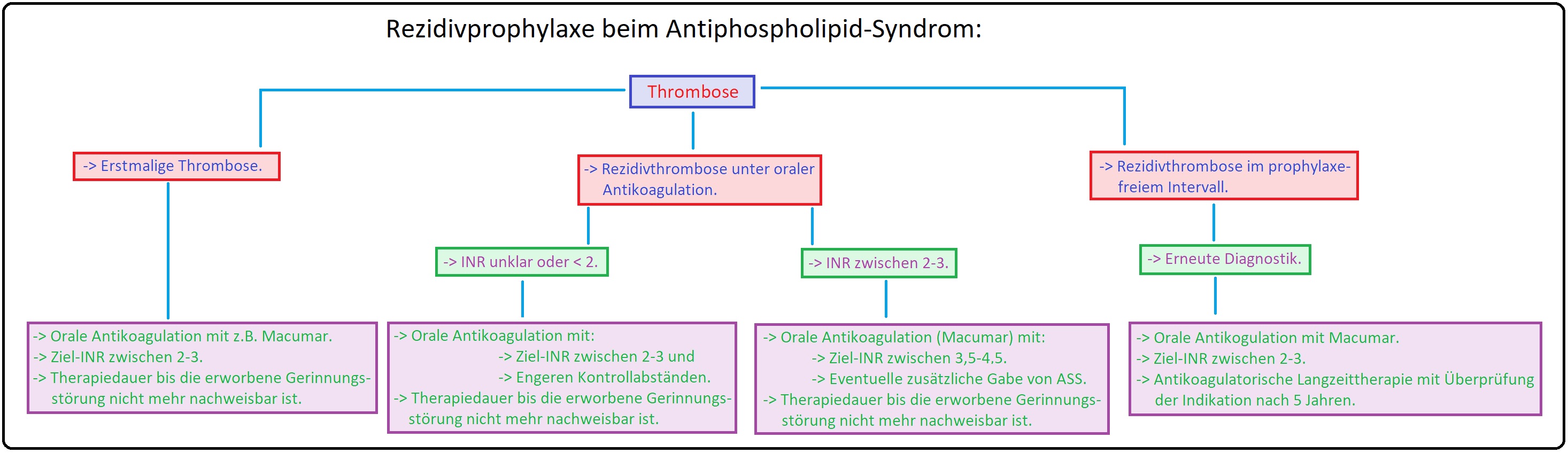
→ IV: Bei habituellen Aborten (APS assoziiert) wird eine langfristige Kombinationstherapie aus ASS und niedermolekularem Heparin in niedriger Dosierung appliziert.
→ V: Catastrophic APS: Liegt diese schwere Verlaufsform vor und sind mehr als 3 Organsysteme betroffen werden Plasmapherese, hochdosierte Glukokortikoide, hochdosierte (intravenöse) Immunglobuline sowie Cyclophosphamid eingesetzt.