→ Definition: Beim von-Willebrand-Jürgens-Syndrom handelt es sich um eine zumeist autosomal-dominante vererbte Störung des vW-Faktors, die zu einer kombinierten thrombozytären und plasmatischen Gerinnungsstörung führt. Die vermehrte Blutungsneigung entsteht aufgrund des Ausfalls der vWF-Funktion in zweierlei Hinsicht:
→ I: Die Bindung und Stabilisierung des Faktor VIII und
→ II: Die Bindung der Plättchen an das Endothel ist nicht mehr gewährleistet.
→ Epidemiologie: Das Von-Willebrandt-Syndrom stellt die häufigste hereditäre Gerinnungsstörung dar (Gesamtprävalenz 10/100000 Einwohnern), die beide Geschlechter gleichermaßen betreffen kann.
→ I: Die Prävalenz symptomatischer Patienten liegt bei 1,25/10000 Einwohner und
→ II: Die Prävalenz für asymptomatische Fälle liegt bei etwa 1%.
→ Ätiopathogenese:
→ I: Morphologie: Das vWF-Monomer hat ein Molekulargewicht von 309000 Dalton und besitzt genau wie der Faktor VIII drei A-, zwei B- und eine C-Domäne sowie zusätzlich noch vier D-Domänen. Das Gen des vW-Faktors ist auf Chromosom 12 lokalisiert und er wird überwiegend in Endothelzellen und Megakaryozyten synthetisiert. Die Vorstufe, der Pro-Willebrand-Faktor, bildet initial ein Dimer und anschließend Multimere, die in den Weibel-Palade-Bodies (= zytoplasmatische Organellen von Endothelzellen) gespeichert werden. Der von Willebrand-Faktor wird durch Abspaltung des Propeptids freigesetzt. Das vWF-Protein besitzt Bindungsstellen für Faktor VIII, Glykoprotein Ib, IIb und IIIa sowie für Kollagen.
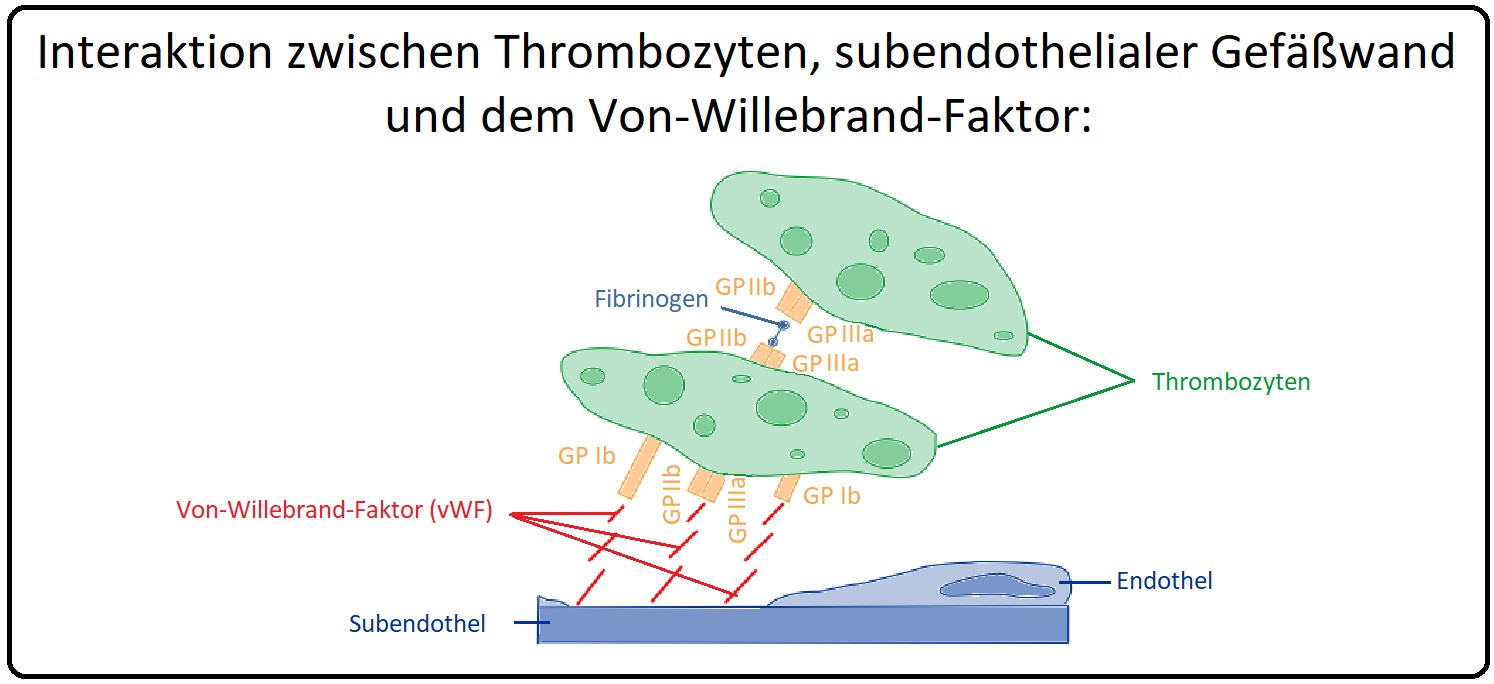
→ II: Beim angeborenen vW-Syndrom können 3 Typen unterschieden werden:
→ 1) Typ I: Stellt mit bis zu 80% der Fälle die häufigste Form dar, wird autosomal-dominant vererbt und ist durch eine quantitative Verminderung des vWF bedingt.
→ 2) Typ II: Diese Form wird ebenfalls autosomal-dominant vererbt und beruht auf einer qualitativen Abnormität des Faktors.
→ 3) Typ III: Wird autosomal-rezessiv vererbt und weist einen deutlich verminderten Spiegel des vWF auf.
→ III: Die Lokalisation des Gendefektes ist insbesondere bei den Typen I und III noch nicht genau bekannt. Beim Typ II wiederum sind einige Punktmutationen bekannt:
→ 1) Subtyp IIa: Es besteht eine Punktmutation in der A2-Domäne. Das betroffene Protein wird durch eine spezifische Plasmaprotease abgebaut (und zirkuliert in verminderter Konzentration).
→ 2) Subtyp IIb: Hierbei zeigt sich eine Punktmutation in der A1-Domäne, die für die GPIb-bindende Domäne kodiert. Folge ist eine ausgeprägte Überaktivität des Proteins mit vermehrter Bindung des mutierten vWF an Plättchen mit konsekutiv vermehrter Elimination (Verlust) des vWF-Plättchen-Komplexes.
→ 3) Subtyp IIn: Hierbei ist eine Mutation am N-terminalen Ende des von-Willebrand-Faktors (= Bindungsstelle des Gerinnungsfaktor VIII). Daraus resultiert eine verminderte Bindungsfähigkeit des vWF für den Faktor VIII, was zum Krankheitsbild der leichten Hämophilie führt.
→ IV: Zudem existiert auch noch ein erworbenes Von-Willebrand-Jürgens-Syndrom; phänotypisch liegt ein Mangel an von-Willebrand-Faktor vor, der für die Funktion des Faktor VIII und Thrombozyten von Bedeutung ist. Ursachen hierfür sind u.a.:
→ 1) Lymphoproliferative Erkrankungen und monoklonale Gammopathie.
→ 2) Maligne Tumoren und Autoimmunerkrankungen.
→ 3) Weitere Ursachen: Sind u.a. Hypothyreose, Medikamente wie Ciprofloxacin und Valproat.
→ Klinisch-relevant: Es existiert eine weitere Klassifikation nach dem Schweregrad des von-Willebrand-Jürgens-Syndrom:

→ Klinik: Das klinische Bild des von-Willebrand-Jürgens-Syndrom ist sehr variabel und reicht von asymptomatischen Verläufen bis hin zum klinischen Bild einer Hämophilie (Schweregrad III).
→ I: Spontane Blutungen wie bei der Hämophilie sind selten, vielmehr manifestieren sich kleiner, diskrete Blutungen wie subkutane Hämatome, Petechien, Schleimhautblutungen, Epitaxis, Neigungen zu Nachblutungen bei Bagatelltraumen, allgemein posttraumatisch und postoperativ (z.B. auch nach Zahnextraktion oder Tonsillektomie, etc.).
→ II: Bei 10% der Betroffenen manifestieren sich Menorrhagien und gastrointestinale Blutungen, insbesondere bei Nachweis von intestinalen Angiodysplasien können diese Blutungen lebensbedrohlich sein.
→ Diagnose: Die Diagnostik des von-Willebrand-Jürgens-Syndroms ist in mehrerer Hinsicht schwierig, da u.a. die Defekte meist leicht sind, es existiert eine große biologische Variabilität bezüglich der Faktor VIII und vW-Faktor-Aktivität, etc. Eine umfangreiche Eigen- und Familienanamnese ist somit obligat.
→ I: Zu den allgemeinen Screeningtests des Von-Willebrand-Jürgens-Syndrom (Abb.: Wichtige Gerinnungsparameter und ihre Normwerte) zählen u.a.:
→ 1) aPTT (= aktivierte partielle Thromboplastinzeit normal oder verlängert).

→ 2) Blutungszeit (verlängert) und
→ 3) PFA (= hierbei handelt es sich um einen Thrombozytenfunktionstest = platelet-function-analysis).
→ Klinisch-relevant: Personen mit der Blutgruppe 0 weisen eine 30%ige niedriger Von-Willebrand-Faktor Aktivität auf als alle anderen.
→ II: Diagnostisch von großer Bedeutung ist die Bestimmung der vWF-Aktivität:
→ 1) Diese wird mit Hilfe des Riskocetin-Cofaktor-Test (Normalwert > 50%) eruiert. Bei Risocetin handelt es sich um ein Antibiotikum, dass in Abhängigkeit von der vWF-Konzentration Thrombozyten aggregiert. Ist die vWF-Aktivität reduziert ist der Test positiv und stellt die Diagnose (eine normale Aktivität schließt das Von-Willebrand-Jürgens-Syndrom weitestgehend aus).
→ 2) Eine Ausnahme bildet die Typ IIn Mutation; hierfür müssen weitere diagnostische Untersuchungen wie z.B. Bestimmung des vWF-Antigens oder Faktor VIII-Aktivität, etc. durchgeführt werden.
→ Differenzialdiagnose: Von dem von-Willebrand-Jügens-Syndrom muss insbesondere die Hämophilie A/B und die Hemmkörperhämophilie abgegrenzt werden. Klinische Unterschiede sind v.a. das Fehlen vom petechialen Blutungstyp sowie keine Verlängerung der Blutungszeit.
→ Therapie:
→ I: Allgemeinmaßnahmen: Bei akuten Blutungen kann ein Druckverband, bei Epitaxis eine Nasentamponade, etc. angelegt werden.
→ II: Spezielle Maßnahmen:
→ 1) Bei Typ I und II (Ausnahme Typ IIb) ist eine Therapie oder Prophylaxe nur bei Operationen, schweren Traumen gastrointestinalen Blutungen indiziert. Die Therapie der ersten Wahl ist die Applikation von Desmopression in einer Dosierung von 0,3-0,4µg/kgKG als Kurzinfusion (alle 12 Stunden über maximal 5 Tage); dies führt zu einem 3-4-fachen Anstieg der Risocetin-Cofaktor- und Faktor-VIII-Aktivität.
→ Klinisch-relevant: Insbesondere bei längerer Applikation besteht die Gefahr der Tachyphylaxie (d.h. zu einem zu geringen oder fehlenden Anstieg von Risocetin-Cofaktor und Faktor-VIII), sodass Prä- und Postinfusionskontrollen obligat sind.
→ 2) Beim Typ IIb und Typ III ist das das Desmopression wirkungslos, sodass eine Indikation für eine Therapie mit Von-Willebrand-Faktor-haltigem Konzentrat indiziert ist. Konzentrate die den vWF enthalten sind Haemate und IMMUNATE, während Faktor-VIII-Konzentrate keinen vWF enthalten und somit nicht für die Therapie geeignet sind.