→ Definition:
→ I: Bei der Riesenzellarteriitis handelt es sich um eine granulomatöse Vaskulitis (Abb.: Klassifikation der Vaskulitiden) der großen Arterien mit Langerhans-Riesenzellen unterschiedlicher Lokalisation; in 2/3 der Fälle ist sie fakultativ mit einer Polymyalgia rheumatica assoziiert:
→ 1) Arteriitis cranialis: Meist im Versorgungsgebiet der A. carotis (A. carotis externa). Sie wird auch Arteriitis temporalis (= Morbus Horton) bezeichnet.
→ 2) Arteriitis extra-cranialis: Mit Beteiligung des Aortenbogens, der Aorta sowie der oberen und unteren Extremität.
→ II: Polymyalgia rheumatica: Die Polymyalgia rheumatika ist eine mit ausgeprägten symmetrischen Schulter- und Beckengürtelmyalgien einhergehende hoch-inflammatorische Erkrankung des höheren Lebensalters.
→ Epidemiologie:
→ I: Die Riesenzellarteriitis stellt die häufigste Vaskulitisform in Westeuropa dar.
→ II: Die Inzidenz liegt bei 15-30/100000/Jahr, wobei Frauen häufiger als Männer betroffen sind (3:1).
→ III: Sie manifestiert sich vor allem im höheren Lebensalter jenseits des 50. Lebensjahrs (und nimmt mit dem Alter noch deutlich zu).
→ Ätiologie: Die Ursache ist bis heute nicht genau bekannt; angenommen wird eine genetische Prädisposition, die durch äußere Umweltfaktoren (Virusinfektion z.B. HBV, Parvoviren, Bakterieninfektion z.B. Mycoplasma pneumoniae, Clamydien etc.) getriggert wird. Es besteht eine vermehrte Assoziation mit HLA-DR4 und -DRB1. Befallen wird insbesondere die
→ I: A. temporalis und
→ II: A. ophthalmica.
→ III: Es können aber auch intrakranielle Arterien einschließlich des Basilarkreislaufes betroffen sein, sodass Symptome eines Schlaganfalls (z.B. Hemiparese) auftreten können.
→ Klinik: Beginnt meist schleichend mit Allgemeinsymptome wie Müdigkeit, Abgeschlagenheit, Muskelschmerzen, Morgensteifigkeit, Gewichtsverlust, Nachtschweiß und Fieber, evtl. entwickelt sich sogar eine Depression.
→ I: Pathognomonisch für die Riesenzellarteriitis ist die Claudiacatio der Kau-, Zugen- und Schlundmukulatur (ist überwiegend ein Vorzeichen des Sehverlustes).
→ II: Arteriitis cranialis: (= Arteriitis temporalis = Horton-Syndrom):
→ 1) Charakterristisch sind ein akut neu auftretender, pochender Schläfenkopfschmerz, Berührungskopfschmerz (eher unilateral), der durch Husten, Kopfbewegungen etc. verstärkt wird, sowie eine Claudicatio masticatoria mit abnormer Ermüdbarkeit und Schmerzen der Kaumuskulatur beim Essen (in 30% der Fälle; pathognomonisch) infolge einer Mangeldurchblutung.
→ 2) Die A. temporalis ist deutlich verhärtet, druckschmerzhaft und es besteht evtl. eine Pulslosigkeit.
→ 3) Augenbeteiligung: Mit Augenbewegungsschmerzen, Doppelbilder bei Befall der Augenmuskulatur bzw. N. abducens Parese, Sehstörungen bis hin zum Visusverlust durch eine anteriore ischämische Opticusneuropathie bzw. Zentralarterienverschluss.
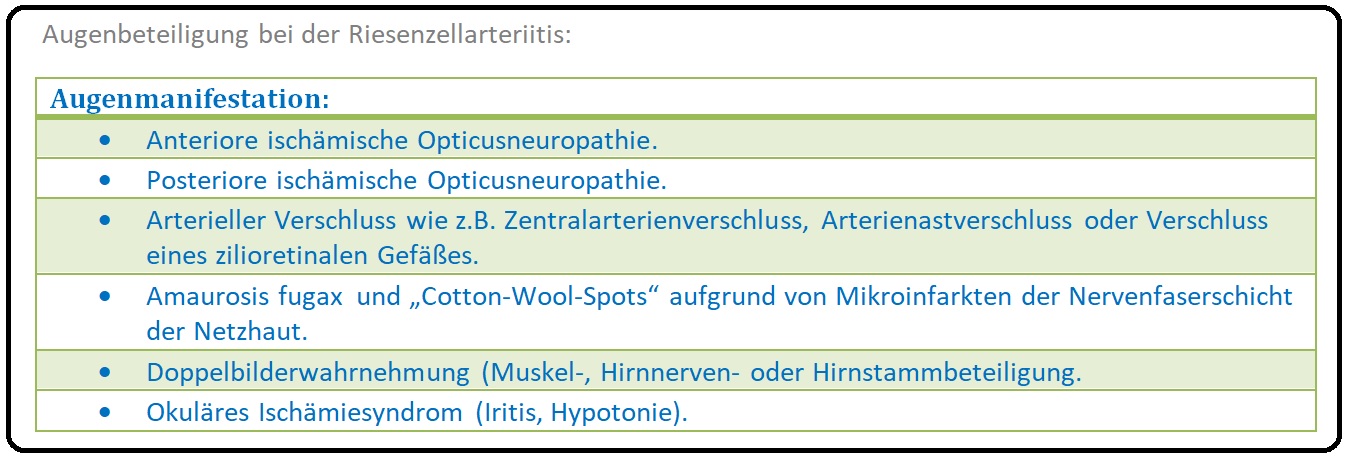
→ III: Arteriits extra-cranialis: Manifestiert sich ein extrakranieller Befall z.B. der Aorta, Koronar- oder der Extremitätenarterien sind u.a. typische klinische Zeichen:
→ 1) Aortenbogensyndrom,
→ 2) Arm-Claudicatio mit Blutdruckseitendifferenz und abgeschwächtem Puls.
→ 3) Thorakale Aortitis mit deutlich erhöhter Inzidenz von Aortenaneurysma und -dissektion.
→ 4) KHK und Myokardinfarkt sowie
→ 5) Häufiger Befall des Posteriorstromgebietes mit möglicher Entwicklung einer TIA und/oder Apoplexie.
→ IV: Polymyalgia rheumatica: Leitsymptome sind ein gürtelförmiger Schmerz oftmals nachts im Bereich des Becken- und Schultergürtels sowie eine Morgensteifigkeit. Des Weiteren kann sich eine Bursitis subdeltoideus bzw. subacromialis entwickeln.
→ Komplikation:
→ I: Ist eine innerhalb von Stunden, sich entwickelnde Blindheit bei Verschluss der A. centralis retina aufgrund einer ischämischen Optikusneuropathie.
→ II: Zerebrale Durchblutungsstörungen mit TIA und Apoplexie etc.
→ Diagnose:
→ I: Anamnese/klinische Untersuchung: Nachweis einer verhärteten und geschlängelten A. temporalis.
→ II: Labor: Deutliche BSG-Erhöhung mit Sturzsenkung (> 40mm/1. Stunde) CRP-Erhöhung und Thrombozytose, evtl. Leukozytose und Anämie (im Vergleich zur Myositis lassen sich keine Auto-AK sowie CK-Erhöhung nachweisen).
→ III: Bildgebende Verfahren:
→ 1) Sonographie: Deutliche echoarme Intima-Media-Verbreiterung mit Lumeneingengung der A. temporalis als Zeichen eines Wandödems bzw. periarteriellen Entzündung (Halo), ein pathologisches Strömungsmuster sowie evtl. Nachweis einer segmentalen Stenosierung.
→ 2) Hochauflösendes Kontrastmittel-MRT: Stellt ein sehr sensitives Verfahren zur Diagnosestellung dar.
→ 3) Biopsie der A. temporalis: Aufgrund des diskontinuierlichen Gefäßbefalls schließt ein negatives Ergebnis eine Riesenzellarteriitis nicht aus. Histologisch zeigt sich eine Infiltration, bestehend aus Monozyten und T-Helfer-(CD4)-Zellen sowie der Nachweis von Granulomen und Langerhans-Riesenzellen.
→ Klinisch-relevant:
→ A) Die Größe des Biopsat-Materials sollte mindestens 2cm betragen, um die Gefahr eines falsch-negativen Ergebnisses zu minimieren.
→ B) Eine Kontraindikation für eine Temporalisbiopsie besteht, wenn sich dopplersonographisch eine Stenosierung der A. carotis interna mit konsekutivem Kollateralkreislauf über die A. carotis externa darstellt.
→ IV: Ophthalmologische Untersuchung: Evtl. Nachweis eines roten Fleckes in der Fovea-centralis bei Verschluss der A. centralis rentinae.
→ V: Diagnosekriterien:
→ 1) Kriterien zur Diagnosestellung einer Polymyalgia rheumatica nach Bird: Es müssen mindestens 3 der 6 nachfolgenden Kriterien zutreffen, damit die Diagnose gestellt werden kann.

→ 2) Diagnosestellung der Arteriitis temporalis nach den ACR-Kriterien (American-College-of-Rheumatology): Es müssen mindestens 3 der 5 Kriterien zu treffen.

→ Differenzialdiagnose: Von der Riesenzellarteriitis bzw. Polymyalgia rheumatica müssen insbesondere nachfolgende Erkrankungen abgegrenzt werden:
→ I: Gesichts- und Kopfschmerzen anderer Genese wie z.B. Trigeminusneuralgie, Migräne, Sinusitis, Meningitis, Enzephalitis, Subarachnoidalblutung, etc.
→ II: Amraurosis fugax bei Verschluss der A. carotis bzw. bei Embolie.
→ III: Myasthenia gravis.
→ IV: Polymyalgia rheumatica:
→ 1) Dermatomyositis/ Polymyositis mit CK-Erhöhung,
→ 2) Rheumatoide Arthritis (late-onset).
→ Therapie:
→ I: Arteriitis cranialis:
→ 1) Es sollte eine sofortige hochdosierte Glukokortikoid-Therapie (aufgrund des guten Ansprechens) mit einer Dosis > 1mg/kgKG/d meist 100mg/d durchgeführt werden.
→ 2) Besteht eine Augensymptomatik (evtl. Amaurosis fugax) ist eine Stoßtherapie mit Prednisolon über 3 Tage indiziert, um ischämische Komplikationen (Erblindung) zu vermeiden.
→ Klinisch-relevant: Ein Augenbefall bei der Riesenzellarteriitis stellt immer eine medizinische Notfallsituation dar. Eine wichtige Sofortmaßnahme ist die Applikation von Methylprednisolon in einer Dosierung von 1000mg/d über 3 Tage. Anschließend wird auf eine orale Gabe umgestellt.
→ 3) Nach klinischer Besserung erfolgt eine langsame Dosisreduktion um 5mg/Woche über Monate bis zu einer Erhaltungsdosis < 7,5mg/d, die aufgrund des hohen Rezidivrisikos über mindestens 24 Monate appliziert wird.
→ II: Bei einer Steroid-Unverträglichkeit oder zu hoher Erhaltungsdosis (oberhalb der Cushingschwelle) kann eine Steroidreduktion durch die Kombination mit einem Immunsuppressiva (z.B. Methotrexat 7,5-15mg/Woche) erreicht werden. Bei Therapieversagen ist die Applikation von Ciclosporin A indiziert.
→ III: Polymyalgia rheumatica: Sie spricht gut auf eine mittel- bis niedrig-dosierte Glukokortikoidtherapie mit einer Initialdosis von 15-20mg/d an.
→ Prognose:
→ I: Unbehandelt führt die Riesenzellarteriitis in 30% zur Erblindung bzw. zu potenziell lebensbedrohlichen Erkrankungen (durch mögliche rebro- und kardiovaskuläre Ereignisse. Bei frühzeitiger und adäquater Therapie jedoch ist die Prognose günstig, da die RZA gut auf die Kortikosteroidtherapie anspricht und komplikationslos ausheilen kann.
→ II: In ca. 30% der Fälle manifesitert sich im weiteren Verlauf ein Rezidiv.
→ III: Prognostisch bedeutend ist die Ausbildung eines Aortenaneurysmas, das bei diesen Patienten bis auf das 17-fache erhöht ist.