→ Definition: Bei der Takayasu Arteriitis handelt es sich um eine (thrombosierende) Vaskulitis, die durch eine granulomatöse Entzündung des Aortenbogens und der abgehenden Gefäße mit konsekutiver Stenosierung (bis hin zur Gefäßokklusion) gekennzeichnet ist. Abhängig vom Typus können Koronar-, Pulmonal- und Nierenarterien mitbetroffen sein (Abb.: Klassifikation der Vaskulitiden).
→ Epidemiologie:
→ I: Die Takayasu-Arteriitis ist mit einer Inzidenz von < 1/100000/Jahr eine sehr seltene Erkrankung in den westlichen Ländern.
→ II: Ein gehäuftes Auftreten findet man in China, Japan, Indien und Afrika.
→ III: Frauen sind deutlich häufiger als Männer (9:1) betroffen, wobei der Manifestationsgipfel vor dem 40. Lebensjahr liegt.
→ Ätiologie:
→ I: Die Ursache der Erkrankung ist noch nicht geklärt.
→ II: Sie stellt eine granulomatöse Arteriitis der Aorta und ihrer Hauptäste dar und weist u.a. Aneurysambildungen und/oder ein Aortenbogensyndrom auf.
→ Pathogenese: Initial besteht eine Entzündung der Adventitia, die mit einer lymphoplasmazellulären Infiltration der Vasa vasorum einhergeht. Im weiteren Krankheitsverlauf, insbesondere in der akuten Phase, kommt es zu einer vermehrten Vaskularisation der Gefäßwand durch Kapillareinsprossung, ausgehend von der Adventitia in die Tunica media und Intima. Diese wird von lymphpasmazelullären Infiltrationen und Riesenzellen begleitet. Folge ist u.a. eine ausgeprägte Verbreiterung der Intima. Mit Abklingen des Entzündungsprozesses werden die Gefäßschichten durch Narbengewebe (= Fibrosierung) ersetzt, was wiederum zu einer deutlichen Zunahme der Aortenwandung führt. Hieraus resultieren Stenosen der Aorta und ihrer großen Gefäßabgänge.
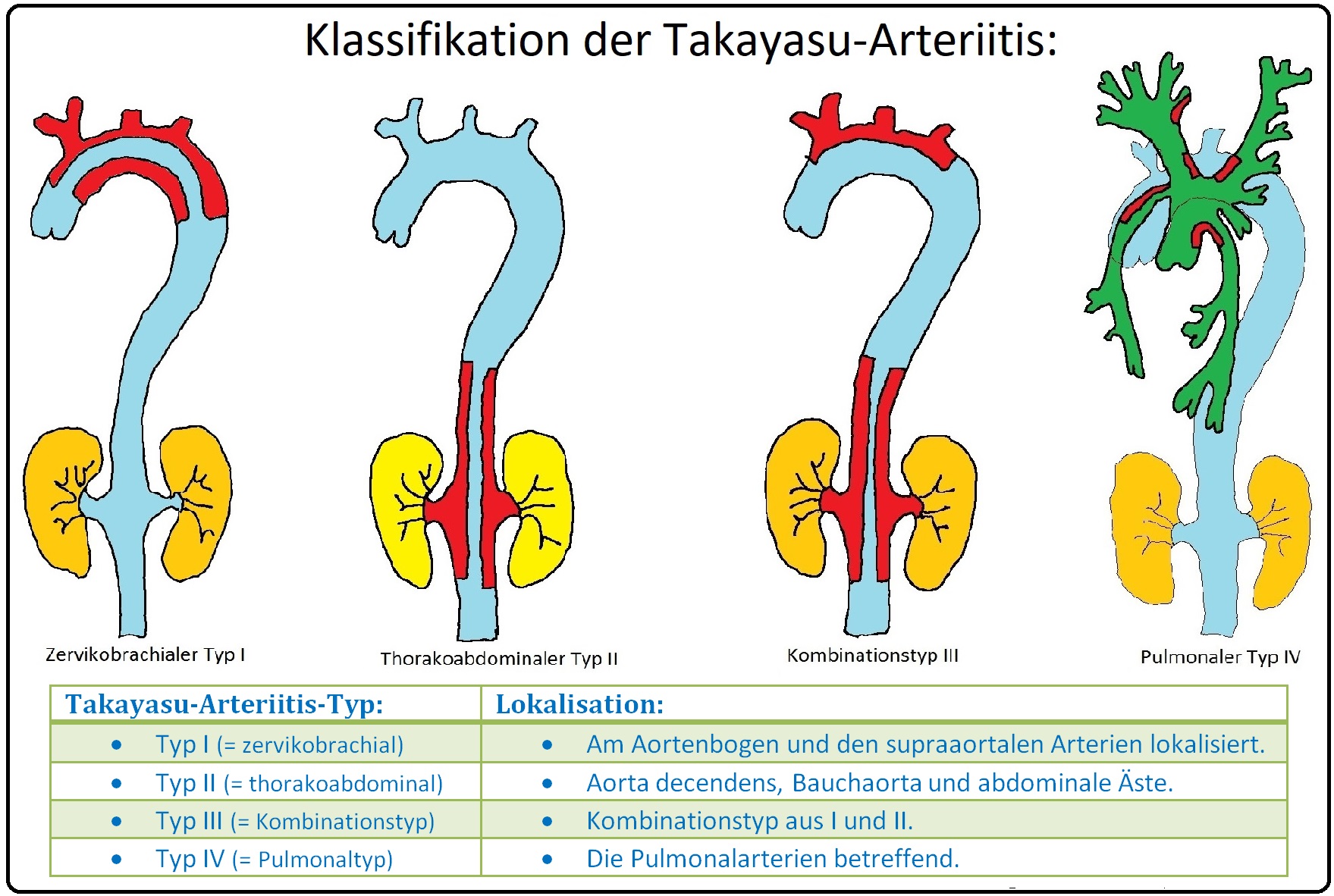
→ Klassifikation: Die Takayasu-Arteriitis wird nach ihrer Lokalisation in verschiedene Subtypen unterteilt. Besonders häufig ist die linke Arteria subclavia betroffen:
→ Klinik: Kardinalbefunde der Takayasu-Arteriitis sind die Seitendifferenz des arteriellen Blutdrucks sowie eine Aorteninsuffizienz:
→ I: Präokklusive Phase: (= Prepulsless Phase) Meist schleichender Beginn mit Allgemeinsymptomen wie Myalgien, Müdigkeit, Abgeschlagenheit, Gewichtsverlust, Nachtschweiß und Fieber über Monate bis z.T. Jahre.
→ II: Okklusive Phase: (= Pulsless-Phase) Hierbei stehen Symptome infolge einer Minderperfusion im Vordergrund.
→ 1) Schwindelattacken, Synkopen, Sehstörungen, Gesichtsfeldausfälle, Aphasie, Hemiparese bis hin zur Apoplexie aufgrund einer zerebralen Minderdurchblutung.
→ 2) Blasse, kalte Haut, Pulsverlust, trophische Hautveränderungen sowie Claudicatio intermittens bis hin zum Subclavian-Steal-Syndrom.
→ 3) Weitere Symptome sind Angina abdominalis, Angina pectoris, renale Hypertonie bei Nierenarterienverschüssen, seltener Erythema nodosum und Raynaud-Syndrom.
→ III: Komplikationen: Wichtige vital-bedrohliche Komplikationen sind der Myokardinfarkt und der Hirninfarkt.
→ Diagnose:
→ I: Anamnese/Klinische Untersuchung: Klassische Zeichen sind abgeschwächter Puls bis -verlust, Blutdruckseitendifferenz von > als 10mmHg (z.T. > 30mmHg) und Strömungsgeräusche über der betroffenen Arterie (insbesondere über den Karotiden und der Aorta abdominalis, seltener über den Femoralgefäßen). Evtl. auskulatatorischer Nachweis einer Aorteninsuffizienz.
→ II: Labor: In der akuten Phase CRP-Erhöhung, die BSG ist deutlich beschleunigt (z.T. > 50mm/h = Sturzsenkung), normochrome Anämie, evtl. leichte Leukozytose sowie Anstieg der Alpha-2-Globuline.
→ III: Bildgebende Verfahren:
→ 1) Sonographie: Nachweis einer konzentrisch verdickten, echoarmen Gefäßwand mit Halo (= Makkaroni-Phänomen) und einer Einenung des Gefäßlumens.
→ 2) CT-/MR-Angiographie: Darstellung von Stenosen und Aneurysma im Verlauf der gesamten Aorta und ihrer Nebenäste.
→ IV: Die Diagnose wird nach den ACR-Kriterien (= American-College of Rheumatologie) gestellt.
→ 1) Alter unter dem 40. Lebensjahr.
→ 2) Einseitige Pulsabschwächung bzw. -verlust.
→ 3) Blutdruckseitendifferenz von > 20mmHg.
→ 4) Störmungsgeräusche über der stenosierten Arterie.
→ 5) Claudicatio intermittens, gerade der oberen Extremität,
→ 6) Pathologischer Angiographiebefund.

→ Differenzialdiagnose: Von der Takayasu-Arteriitis müssen insbesondere nachfolgende Erkrankungen abgegrenzt werden:
→ I: Arteriosklerotische Erkrankungen wie pAVK, KHK und TIA, arteriosklerotisch bedingtes Aortenbogensyndrom,
→ II: Vaskulitis anderer Genese (Abb.: Klassifikation der Vaskulitiden).
→ III: Syphilitische Mesoaortitis und das mykotische bzw. idiopathische thorakale Aortenaneurysma.
→ IV: Arteriitis temporalis bzw. Polymyalgia rheumatica mit Befall des Aortenbogens.
→ Therapie:
→ I: Konservativ:
→ 1) Mittel der Wahl ist die immunsuppressive Therapie mit Glukokortikoiden (z.B. Prednisolon über mindestens 1 Jahr) und/oder Methotrexat als Reservesubstanz.
→ 2) Des Weiteren erhalten die Patienten zur Hemmung der Thrombozytenaggregation mit z.B. Acetylsalicylsäure in der Dosierung von 100mg/d.
→ II: Operativ: Im entzündungsfreien Intervall kann eine interventionelle Therapie (PTA + Stentimplantation) oder eine Bypass-Operation zur Gefäßrekonstruktion indiziert sein.
→ Prognose: Ohne Behandlung verläuft die Takayasu-Arteriitis chronisch progredient mit der Gefahr der Minderperfusion von Herz und Gehirn und charakteristischen klinischen Symptomen wie KHK, Myokardinfarkt, Schlaganfall, etc.