→ Definition: Die Wegener-Granulomatose stellt eine nekrotisierende (ANCA-positive) Vaskulitis der kleineren bis mittelgroßen Gefäße (Abb.: Klassifikation der Vaskulitiden) mit Ausbildung nekrotisierender, nicht-verkäsender Granulome dar. Hierbei sind insbesondere der obere Respirationstrakt und die Nieren, möglicherweise aber auch weitere Organe betroffen.
→ Epidemiologie:
→ I: Die Wegener Granulomatose ist die häufigste ANCA-assoziierte Vaskulitis (betrifft Kapillaren, Venolen, Arteriolen und Arterien), ihre Inzidenz liegt bei ca. 1-5/100000/Jahr.
→ II: Die Erkrankung manifestiert sich zumeist zwischen dem 40.-50. Lebensjahr (kann aber in jedem Lebensalter auftreten), wobei Frauen genauso häufig wie Männer betroffen sind.
→ Ätiologie: Die Pathogenese des Morbus Wegener ist nicht genau bekannt es wird jedoch die Einwirkung des proinflammatorischen Zytokins TNF-alpha, welches eine Translokation des PR3-Gens aus dem Zytoplasma an die Granulozytenoberfläche induziert vermutet. In diesem Zusammenhang kann cANCA an das PR3-Antigen binden und eine Aktivierung der Granulozyten hervorrufen. Folge ist die Freisetzung lysosomaler Enzyme und O2-Radikale, die das Gefäßendothel destruieren.
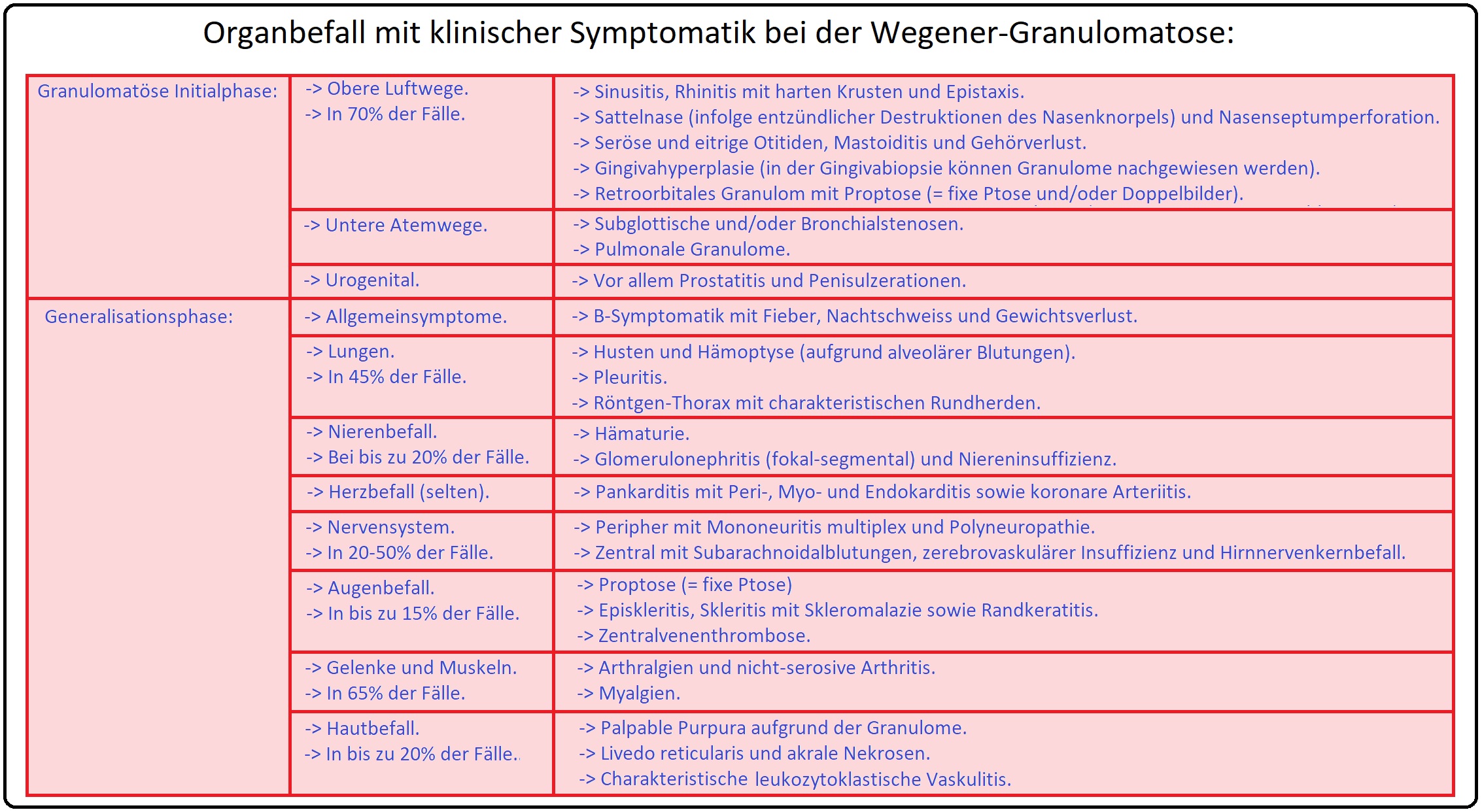
→ Klinik: Klinisch können insgesamt 5 Stadien unterschieden werden:
→ I: Lokalisiertes Stadium: (= Symptomarme Initialphase, die über Monate bis Jahre verlaufen kann). Betrifft typischerweise den Hals-Nasen-Ohrenbereich, Augen sowie den oberen Respirationstrakt. Charakteristische Symptome sind u.a. im Augenbereich Konjunktivitis (bis hin zur Scleromalacia perforans), Keratitis und venösen Abflussbehinderungen, eine chronische Rhinitis/Sinusitis mit blutig-borkigem Schnupfen, eine Ostitis media, evtl. eine Nasenscheidewandperforation; des Weiteren können sich ein ulzerierender Oropharynx und Lungenherde mit möglichen Einschmelzungen (= sogenannte Pseudokavernen) manifestieren.
→ II: Frühgeneralisationsstadium: Beinhaltet jede Art der Manifestation ohne Vital- oder Organbedrohung (u.a. unspezifische Allgemeinsymptome wie Gewichtsverlust, Nachtschweiß und Fieber).
→ III: Generalisationsstadium: Hierbei findet man eine Beteiligung der Niere (Serumkreatinin < 500µmol/l = 5,6mg/dl) oder eine andere Organbedrohung. Alveoläre Hämorrhagien mit Hämotypsen in Kombination mit einer rapid-progressiven Glomerulonephritis (und nephritischen Syndrom) wird als pulmorenales Syndrom bezeichnet. Weitere Symptome können Episkleritis, Keratokonjunkivitis, Arthralgien, Polyarthritis, Myalgien, periphere Neuropathien, ZNS-Symptome (Hirnnervenlähmungen, epileptiforme Anfälle, Apoplexie), Hautsymptome (z.B. Petechien, palpable Purpura, Ulzerationen etc.) sowie Beteiligung des Herzens mit Entzündung der Kornargefäße, Valvulitis oder Perikarditis sein.
→ IV: Schweres lebensbedrohliches Generalisationsstadium: Charakteristisch ist das Nierenversagen (Serumkreatinin > 500µmol/l; 5,6mg/dl) oder das Versagen eines anderen Organs wie der Lunge mit Intubationspflichtigkeit innerhalb von Stunden.
→ V: Refraktäres Stadium: Es handelt sich um eine progressive, Steroiden gegenüber refraktäre Erkrankung.
→ Diagnose:
→ I: Labor:
→ 1) BSG- und CRP-Erhöhung, Leukozytose (dienen auch zur Aktivitätsbeurteilung), Thrombozytose und Anämie,
→ 2) Kontrolle der Retentionsparameter, der Proteinausscheidung und einer möglichen Erythrozyturie zur Überwachung der Nierenfunktion. Im Urinsediment können möglicherweise dysmorphe Erythrozyten sowie Erhythrozyten- und Leukozytenzylinder nachweisbar sein. (Besteht eine Mirkohämaturie ist die Mitbetimmung der Urinsedimente obligat, um frühzeitig eine Glomerulonephritis zu erkennen).
→ Klinisch-relevant: Der Nachweis von antineutrophilen-cytoplasmatischen-AK (mit zytoplasmatischem Floureszenzmuster) ist pathognomonisch und richtet sich gegen die Proteinase 3; auch ist die Bestimmung des PR3-Titers von Bedeutung, so ist bei einer Verdopplung des PR3-Titers von einem drohenden Schub auszugehen und eine entsprechende Therapieintensivierung einzuleiten.
→ II: Radiologie:
→ 1) Röntgen: Der Nasennebenhöhlen mit Verschattungen; der Lunge mit Nachweis von Infiltrationen die sich als Rundherden darstellen. Pseudokavernen zeigen sich dann im Rahmen von Einschmelzung. Besteht eine alveoläre Hämorrhagie manifestiert sich eine "weiße Lunge".
→ 2) CT/MRT: Der Nasennebenhöhlen/Schädels mit Nachweis von Granulomen , evtl. zerebrale Läsionen.
→ 3) CT-Angiographie: Nachweis von Mikroaneurysmen der Nierengefäße
→ III: Biopsie: Des betroffenen Organs. Charakteristisch ist das histologische Trias bestehend aus:
→ 1) Granulomen,
→ 2) Vaskulitis und
→ 3) Einer Glomerulonephritis.
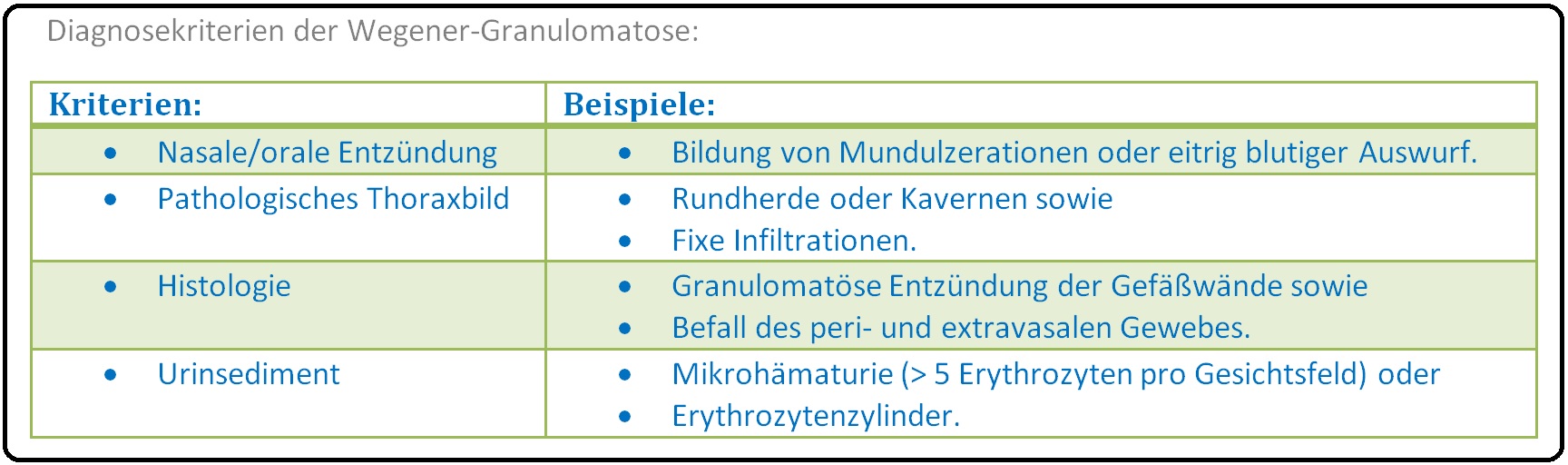
→ Klinisch-relevant: Um die Diagnose der Wegener-Granulomatose zu stellen, müssen mindestens 2 der folgenden 4 ACR-Kriterien zutreffen (Sie weisen eine hohe Sensitivität > 88% und Spezifität > 92% auf):
→ Differenzialdiagnose: Von der Wegener Granulomatose müssen insbesondere nachfolgende Erkrankungen abgegrenzt werden. Hierzu zählen:
→ I: Weitere Kleingefäßvaskulitiden: Wie die
→ 1) Mikroskopische Polyangiitis (Granulome fehlen, pANCA) oder das
→ 2) Churg-Strauss-Syndrom mit allergisches Asthma bronchiale; nur selten ist die Niere beteiligt (Abb.: Klassifikation der Vaskulitiden).
→ II: Goodpasture-Syndrom: Insbesondere bei bestehendem pulmorenalem Syndrom. Hierbei zeigt sich immunhistochemisch eine granuläre Anbalgerung von Immunkomplexen an der glomerulären Basalmembran.
→ Therapie: Die Therapie der Wegener-Granulomatose ist Stadien-abhängig:
→ I: Lokalisiertes Stadium: Bei der Induktionstherapie handelt es sich um eine Kombinationstherapie, bestehend aus Cyclophosphamid 2mg/kgKG (bzw. Methotrexat) und einem Glukokortikoid (z.B. Prenisolon 1-2mg/kgKG) Anschließend erfolgt eine Erhaltungstherapie mit Azathioprin (niedrig dosiert) oder eine Kombinationstherapie aus Methotrexat, Trimethoprim und Sulfamethoxazol.
→ II: Frühsystemisches Stadium: Auch hier existiert eine Induktionstherapie mit einem Steroid + Methotrexat oder Cyclophosphamid. Die Erhaltungstherapie erfolgt mit einem niedrig-dosierten Steroid + Azathioprin oder Methotrexat.
→ III: Generalisationsstadium: Hierbei ist die Applikation von Prednisolon 1mg/kgKG/d indiziert. Anschließend ist die langsame Dosisreduktion auf 10-15mg/d nach Remission obligat.
→ IV: Lebensbedrohliches Generalisationsstadium: Prednisolon-Bolustherapie i.v. über 3 Tage in Kombination mit Cyclophosphamid i.v. sowie eine Zusatztherapie:
→ 1) Einsatz einer Plasmapherese (erwies sich als vorteilhaft).
→ 2) Blasenschutzmittel wie Uromitexan und antifugale Therapie sowie
→ 3) Magenschutz.
→ V: Reservemittel der Wegener Granulomatose sind vor allem Rituximab und Infliximab.
→ Prognose: Unbehandelt schreitet die Wegener Granulomatose rasant voran und die mittlere Überlebensrate beträgt ca. 1 Jahr. Mit Hilfe der immunsuppressiven Therapie erreicht man in 90% der Fälle eine Remission, jedoch besteht ein hohes Rezidivrisiko.