→ Definition: Bei der Hämochromatose handelt es sich um eine hereditäre Stoffwechselerkrankung der Leber (Eisenspeicherkrankheit) mit deutlich erhöhter Eisenaufnahme und -ablagerung in den Parenchymzellen der verschiedenen Geweben wie Leber, Pankreas, Myokard, Hypophyse und Haut.
→ Klassifikation: Nach der Entstehung wird zwischen 2 verschiedene Subtypen der Eisenspeicherkrankheit differenziert:
→ I: Primäre Hämochromatose: Die zumeist autosomal-rezessiv vererbte Punktmutation auf Chromosom 16 (C282Y-Mutation des HFE-Gens) weist 5 verschiedene Subtypen auf und ist durch eine Symptomtrias bestehend aus Diabetes mellitus, Leberzirrhose und Hyperpigemtierung der Haut charakterisiert. Des Weiteren sind exzessive Eisenablagerungen in anderen Organen nachweisbar.

→ II: Sekundäre (erworbene) Hämosiderose: Sie stellt eine erworbene Form dar und wird durch eine Hämosiderose (= Eisenüberlastung ohne Gewebeschädigung) bei z.B. Transfusionsbehandlungen, Anämie, Hämolyse (Thalassaemia major, Porphyria cutanea tarda) oder hämatologischen Erkrankungen wie das myelodysplastische Syndrom oder der Myelofibrose hervorgerufen.
→ Epidemiologie:
→ I: Die homozygote Form der Hämochromatose liegt in Deutschland bei ca. 1/4000 Einwohnern vor, die heterozygote bei 1/20. Es existieren 5 verschiedene, genetische Defekte für den Bronzediabetes, wobei die Mutation des HFE-Gens an Position C282Y (Typ 1= adulte Hämochromatose) die häufigste Form darstellt.
→ II: Männer sind 10x häufiger vom manifesten Typus betroffen als Frauen (vor der Menopause). Dieser Unterschied gleicht sich nach der Menopause aus, da die Menstruation bei der Frau als natürlicher Aderlass fungiert und zu einer Korrektur der Eisenüberlastung führt. Die Diagnosestellung erfolgt meist zwischen dem 40.-60. Lebensjahr.
→ Ätiopathogenese: Die hereditäre Hämochromatose wird durch eine Mutation auf dem HFE-Gen auf dem Chromosom 16 in enger Beziehung zur HLA-Region (75% der Patienten sind HLA-A3 positiv) verursacht. Die beiden bekanntesten Mutationen des HFE-Gens sind C282Y (bei dieser Mutation kommt es zum Austausch von Tyrosin durch Cystein an Position 282 des HFE-Gens; in 90% der Fälle sind Patienten mit Hämochromatose homozygot für die C282Y-Mutation) und H63D. Pathogenetisch besteht eine sowohl eine zelluläre als auch systemische Störung der Eisenhämostase aufgrund einer:
→ I: Massiven enteralen Eisenresorption: Bei Patienten mit Hämochromatose besteht eine vermehrte enterale Aufnahme von Eisen um das 2-4-fache der Norm, sodass es über Jahre zu einer Akkumulation von Fe auf 20-40g (normal 2-3g) im Organismus kommt.
→ II: Vermehrte Eisenabgabe der Makrophagen: Histologisches Kennzeichen der Hämochromatose ist eine massive Eisenspeicherung in den Hepatozyten bei (Eisen-) Aussparung der Makrophagen (= Kupffersche Sternzellen). Pathophysiologisch wird beim Abbau des Hämoglobins das gewonnene Eisen von den Makrophagen (und Erythrozyten) unreguliert an das Blut abgegeben.
→ III: Folge der Eisenüberlagerung: Bei der Eisenüberlagerung in den Zellen wird die Bindungskapazität der Speicherproteine überschritten und ionisiertes Fe akkumuliert mit z.T. schwerwiegenden Folgen:
→ 1) Zellschädigung und -nekrosen.
→ 2) Zunahme der extrazellulären Matrix mit konsekutiver Fibrogenese und
→ 3) Deutlich erhöhtes Risiko für maligne Transformationen.
→ Klinik: Die klinische Symptomatik entwickelt sich insbesondere in Abhängigkeit mit der Eisenüberladung in den verschiedenen Geweben und nimmt im Alter zu. Folgende Organsysteme sind bei der Hämochromatose betroffen. Initial können sich Frühsymptome herauskristallisieren:

Das charakteristische, klinische Bild der Hämochromatose wird jedoch durch die Schädigung der verschiedenen Organsysteme hervorgerufen; Neben Allgemeinsymptomen wie Schwäche, Müdigkeit und Abgeschlagenheit zählen zudem v.a.:
→ I: Leber: Charakteristisch sind Hepatomegalie, evtl. Splenomegalie undim Spätstadium die Leberzirrhose mit der Gefahr der Entwicklung eines hepatozellulären Karzinoms. Sie tritt bei Eisengehalten > 280µmol/g Trockengewicht auf.
→ II: Pankreas: Häufiger Nachweis einer endokrinen Funktionsstörung mit verminderten Glukosetoleranz, die im weiteren Krankheitsverlauf nicht selten in einen Diabetes mellitus übergeht. Ursache hierbei ist nicht die Destruktion der Inselzellen, sondern vielmehr eine Insulinresistenz.
→ III: Haut : Das typische, bronzefarbene Hautkolorit ist nur selten nachweisbar, häufiger jedoch zeigt sich eine graubraune Verfärbung insbesondere in den lichtexponierten Regionen im Genitalbereich, Achselhöhlen und der Mundschleimhaut aufgrund einer Hyperpigemntierung (Melaninzunahme).
→ IV: Endokrine Organe: Entwicklung eines Hypogonadismus mit konsekutivem Libidoverlust und Impotenz aufgrund von Eisenablagerungen in den gonadotropen Zellen der Hypophyse. Bei jungen Frauen mit massiver Eisenablagerung, die nicht durch die Menstruation kompensiert werden kann, manifestiert sich in diesem Zusammenhang eine Amenorrhoe. Auch kann es zu einer Destruktion der Nebennierenrinde kommen.
→ V: Herz: Wichtige kardiale Symptome der Hämochromatose aufgrund von Nekrosen und Fibrosierung sind u.a.:
→ 1) Herzrhythmusstörungen insbesondere die Tachyarrhythmien,
→ 2) Dilatative Kardiomyopathie und
→ 3) Die Herzinsuffizienz.
→ VI: Gelenke: Die Veränderungen an den Gelenken stellt ein Frühsymptom der Hämochromatose dar. Typisch ist hierbei eine chronische Arthritis am 2.-3. Metakarpophalangealgelenk oder an den Interphalangealgelenken mit konsekutiven Schmerzen. Des Weiteren können aber auch das Ellenbogen-, Hüft- oder Kniegelenk betroffen sein. Röntgenologisch zeigt sich eine Verschmälerung des Gelenkspaltes, subchondrale Zysten und im fortgeschrittenen Stadium eine Chondrokalzinose.
→ Komplikation: Eine schwerwiegende Komplikation der Hämochromatose ist die Ausbildung eines hepatozellulären Karzinoms; insofern sind regelmäßige Kontrolluntersuchungen in Zeitintervallen von 6 Monaten zur Früherkennung obligat.
→ Diagnose:
→ I: Anamnese und klinische Untersuchung.
→ II: Labor: Wichtige Parameter sind die:
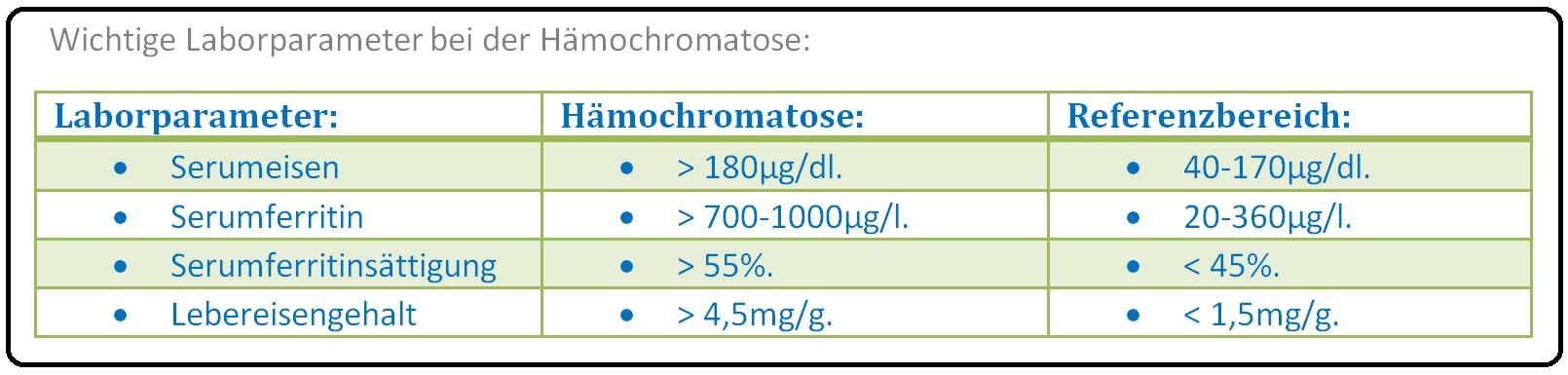
→ 1) Erhöhung des Serumferritins (bei Frauen > 250ng/l und Männern > 300ng/l) sowie die
→ 2) Transferrinsättigung über 60% (bei Frauen > 45%, bei Männern > 55%), die man aus dem Quotienten aus Serumeisen und Serumtransferrin x einem Faktor errechnet wird.
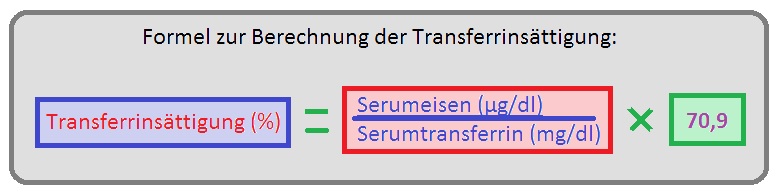
→ 3) Nicht selten ist eine Erhöhung des Serumeisens (> 180µg/dl) und des Ferritins (> 1000µg/l; Hinweis auf Eisenüberladung) eruierbar.
→ 4) HFE-Gendiagnostik: Ein normaler HFE Genotyp schließt eine Hämochromatose nicht sicher aus; vielmehr muss die Gesamtheit aus Klinik, Laborparameter und Gendiagnostik im Zusammenhang gesehen werden.
→ 5) Weitere Laborparameter: Zur Beurteilung der Leberfunktion sind die Transaminasen und bei Verdacht auf ein Leberzellkarzinom das Alpha-Fetoprotein zu bestimmen.
→ III: Leberbiopsie: Mit Histologie und Bestimmung des Lebereisengehaltes ist insbesondere bei Patienten mit Serumferritin > 1000µg/l, erhöhter Transaminaseaktivität und Transferrinsättigung (> 50-60%) gerechtfertigt. Hierbei zeigt sich ein deutlich erhöhter Eisengehalt im Lebertrockengewicht > 4,5 mg/g (= > 80µmol/g; Lebereisen-Index = µmol/g Trockengewicht/Lebensalter > 2,0).
→ Differenzialdiagnose: Von der hereditären Hämochromatose müssen insbesondere die sekundären Siderosen abgegrenzt werden; hierzu zählen:
→ I: Sekundäre Eisenspeicherkrankheiten: (= Siderosen) bei:
→ 1) Hämolytischer Anämie wie die Thalassämia major, hereditäre Sphärozytose, etc., aber auch bei vermehrten Bluttransfusionen oder den myelodysplastischen Syndromen.
→ 2) Alkoholbedingte Siderose durch vermehrte Eisenaufnahme bei chronischer Alkoholabhängigkeit.
→ 3) Siderosen bei chronischen Lebererkrankungen.
→ II: Erhöhte Ferritinwerte bei Entzündungen, da Ferritin ein Akutphaseprotein ist.
→ III: Nicht alkoholische Fettleberkrankheit / NASH.
→ Therapie: Ziel der Behandlung ist die Reduktion des Körpereisens durch u.a.:
→ I: Allgemeinmaßnahmen:
→ 1) Eisenarme Diät durch Meiden von Fleisch und
→ 2) Erhöhte Eisenausscheidung: Durch Aderlässe (stellt die Therapie der Wahl dar). Hierbei werden in der Initialphase 400-500l/Woche (entspricht einem Eisenentzug von 200-250mg) Blut entnommen. Die Dauer der Behandlung richtet sich nach dem Grad der Eisenüberlastung. Das Therapieende wird bei einem Ferritinwert < 50mg/dl und einer Transferrinsättigung < 20% eingeleitet. Es erfolgt zumeist nach mehreren Wochen bis einigen Monaten; nur bei schwerwiegender Eisenüberladungen kann es auch über 1 Jahr andauern. Im Anschluss folgt die Erhaltungtherapie bei der in Zeitabständen von 1-3 Monaten ein Aderlass durch geführt wird.
→ Klinisch-relevant: Die Aderlassbehandlung führt zu einer deutlichen Besserung evtl. auch Normalisierung der Lebergröße, Transaminasen, aber auch der Hautpigmentierung, Kardiomyopathie und des Allgemeinzustandes. Weniger Einfluss hat sie auf den Diabetes mellitus, eine bestehende Leberinsuffizienz und die Arthropathie.
→ II: Medikamentöse Therapie: Eisenchelatbildner wie das Deferoxamin (Deferoxamin kontinuierlich s.c. oder i.v. über Pumpe 20-60mg/kg KG/d; Deferasirox per os 10-40 mg/kgKG/d) sind insbesondere bei Kontraindikationen gegen den Aderlass und der juvenilen Hämochromatose indiziert; weisen jedoch toxische Nebenwirkungen wie Tinnitus, Innenohrschwerhörigkeit, Seh-Störungen insbesondere des Farbsehens durch Retinadestruktionen sowie neurologische Symptome auf.
→ III: Bei bestehender Leberzirrhose HCC-Screening mittels Sonographie und AFP-Bestimmung alle 6 Monate.
→ Klinisch-relevant: Eine Lebertransplantation als ultima ratio ist bei der Hämochromatose nicht geeignet, da der primäre Defekt nicht in der Leber, sondern vielmehr im Darm lokalisiert ist und es somit in der gesunden, transplantierten Leber erneut zur Eisenüberladung kommt.
→ Prognose: Mit der Einführung der Aderlasstherapie ist die Prognose von Patienten in einem präzirrhotischen Stadium sehr gut. Oftmals besteht hierbei eine normale Lebenserwartung.