→ Definition: Bei der primär sklerosierenden Cholangitis handelt es sich um eine idiopathisch fibrosierende Entzündung der intra- und extrahepatischen Gallengänge (diffus oder fokal) mit konsekutiver chronischer Cholestase aufgrund von segementalen (unregelmäßigen) Stenosierungen und Obliterationen. Sind nur sehr kleine intrahepatische Gallengänge der Portalfelder betroffen spricht man von einer "small-duct" PSC mit günstigerer Prognose. Folgen sind rezidivierende bakterielle Infektionen und die Gefahr der Ausbildung einer sekundären biliären Zirrhose.
→ Epidemiologie:
→ I: Bei der primär sklerosierenden Cholangitis handelt es sich um eine seltene Erkrankung; die Inzidenz liegt 1-5 Erkrankungen/100000/Jahr.
→ II: Männer sind doppelt so häufig wie Frauen betroffen, wobei der Manifestationsgipfel zwischen dem 30.-50. Lebensjahr liegt. Die PSC kann jedoch auch im Kindesalter auftreten; hier zeigt sich dann zumeist eine deutlich stärkere entzündliche Aktivität.
→ Ätiologie:
→ I: Die Entstehung der PSC ist bis heute noch nicht genau geklärt; jedoch ist sie vermehrt mit der Colitis ulcerosa (in 70-80% der Fälle, seltener mit dem Morbus Crohn) der Zöliakie, Pankreatitis, Riedel-Struma, Sarkoidose und dem Sjögren-Syndrom (seltener) vergesellschaftet. Diskutiert werden insbesondere genetische sowie exogene Faktoren; eine familiäre Häufung existiert.
→ II: Des Weiteren kristallisiert sich eine HLA-B8 und DR2, DR3 und DR4 Assoziation heraus (nicht selten lassen sich auch Antikörper gegen zytoplasmatische Antigene der neutrophilen Granulozyten = ANCA nachweisen).
→ Klinik: Die Erkrankung beginnt zumeist schleichend. Im Frühstadium sind meist keine Symptome vorhanden, evtl. bestehen erhöhte yGT-Werte im Labor. Erst im weiteren Krankheitsverlauf entwickelt sich eine typische klinische Symptomatik mit:
→ I: Müdigkeit, Abgeschlagenheit, unklare abdominale Schmerzen sowie eine Hepatomegalie.
→ II: Quälender Juckreiz, der zumeist Jahre vor dem cholestatischen Stadium (Cholestase) auftritt; im weiteren Krankheitsverlauf entwickelt sich ein Ikterus sowie Zeichen einer Maldigestion mit Vitaminmangelerscheinungen, Gewichtsverlust und Steatorrhoe.
→ III: Rezidivierende Cholangitiden infolge aszendierender Infektionen mit der klassischen Charcot-Trias, bestehend aus epigastrischen Schmerzen, Ikterus und Fieber.
→ Komplikationen:
→ I: Ausbildung von Gallensteinen, rezidivierende bakterielle Cholangitiden mit der Gefahr der Entwicklung von Leberabszessen.
→ II: Biliäre Zirrhose mit charakteristischen Folgen:
→ 1) Aszites,
→ 2) Hepatorenales Syndrom,
→ 3) Varizenblutungen,
→ 4) Portale Hypertension,
→ 5) Hepatische Enzephalopathie.
→ III: Erhöhtes Risiko für die Entwicklung eines Cholangiozellulären-Karzinoms (CCC), aber auch für das kolorektale Ca. und das Pankreaskarzinom.
→ Diagnose:
→ I: Anamnese/klinische Untersuchung: Bei unklarer cholestatischer Leberwerterhöhung sollte nach Durchfällen und Bauchschmerzen als Hinweis für eine chronisch entzündliche Darmerkrankungen eruiert werden. Des Weiteren können sich eine Druckschmerzhaftigkeit unter dem rechten Rippenbogen, Leberhautzeichen, etc. darstellen.
→ II: Labor:
→ 1) Cholestaseparameter: Zeichen einer Cholestase mit erhöhter AP-, yGT- und LAP-Konzentration. Aber auch die Transaminasen sowie das Bilirubin sind deutlich erhöht. Eine kontinuierlich Zunahme der Hyperbilirubinämie weist auf eine Progredienz und schlechte Prognose hin.
→ 2) Es ist eine Gammaglobulinämie vom IgM-Typ eruierbar.
→ 3) Autoantikörper: Bei der PSC bestehen keine spezifischen Auto-AK, jedoch sind in bis zu 60% die antineutrophilen-cytoplasmatischen AK mit perinukleärer Fluoreszenz (pANCA) nachweisbar.
→ 4) Eine Erniedrigung des Quick-Wertes und Albumins weist auf ein fortgeschrittenes Stadium hin.
→ III: ERCP/MRCP: Sehr sensitiv mit Nachweis von perlenschnurartig-aussehenden Gallengängen durch diffus auftretende Strikturen bzw. Stenosen und Dilatationen. Die nachgewiesenen Strikturen können nach ihrem Stenosierungsgrad unterteilt werden:

→ IV: Leberhistologie: Charakteristische periduktale Fibrosierung durch zwiebelschalenförmige Ummauerung der Gallengänge.

→ Differenzialdiagnose: Von der primär-sklerosierende Cholangitis sind insbesondere nachfolgende Erkrankungen abzugrenzen. Hierzu zählen:
→ I: Cholestase anderer Genese wie Choledocholithiasis, Cholezystitis, Cholangitis, Caroli-Syndrom etc.
→ II: Pruritus anderer Genese,
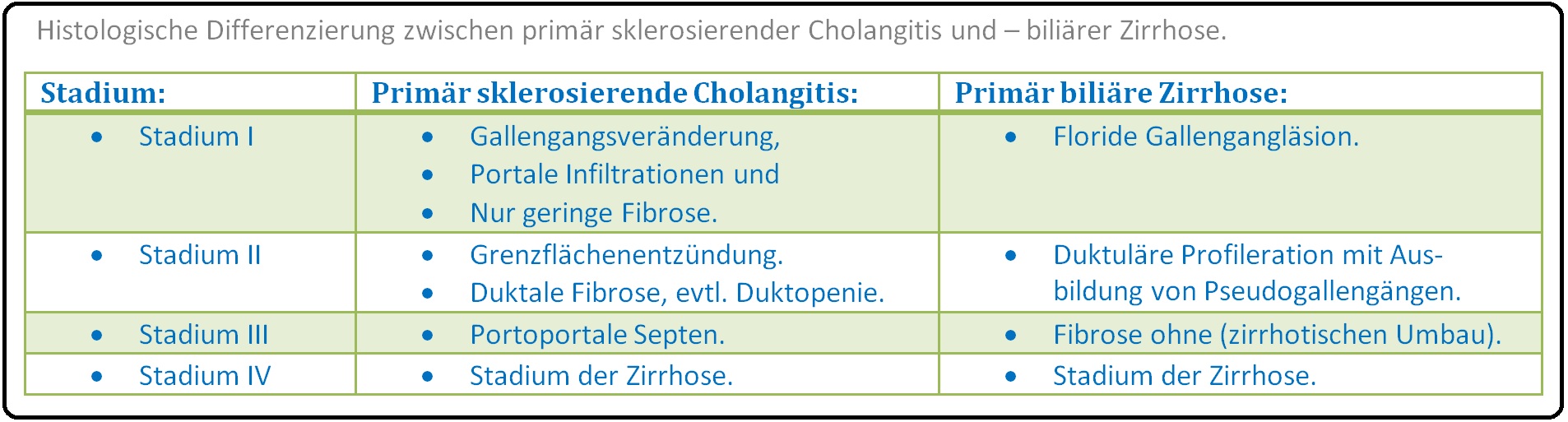
→ III: Weitere Erkrankungen wie die primär biliäre Zirrhose und das cholangiozelluläre Karzinom.
→ IV: Überlappungssyndrom mit der Autoimmunhepatitis in 6% der Fälle.
→ Therapie:
→ I: Medikamentöse Therapie:
→ 1) Es ist eine medikamentöse Therapie mit Ursodesoxycholsäure indiziert. Die Tagesdosis liegt zwischen 15-25mg/kgKG/d.
→ 2) Bei Gallengangsinfektionen wird eine antibiotische Therapie mittels Ciprofloxacin oder Ceftriaxon verabreicht.
→ 3) Die Gabe von Colestyramin kann den quälenden Pruritus deutlich reduzieren
→ II: Invasiv:
→ 1) Bei Stenosierung PTC (perkutane-transhepatische Cholangiographie) mit Ballondilatation und Einlage eine Stents.
→ 2) Ultima ratio bei beginnender Dekompensation der Leberzirrhose ist die Lebertransplantation. Sie stellt die einzige kurative Behandlung dar.
→ Prognose:
→ I: Bei der PSC besteht bis heute keine Heilung, sodass die Prognose ernst ist; die mittlere Überlebenszeit nach Diagnosestellung ohne Lebertransplantation liegt zwischen 10-20 Jahren.
→ II: Da bei der primär-sklerosierenden Cholangitis und der Colitis ulcerosa die Gefahr eines kolorektalen Karzinoms erhöht ist, sollten regelmäßige endoskopische Vorsorgeuntersuchungen erfolgen.