→ Definition: Beim cholangiozellulären Karzinom handelt es sich um eine maligne Neoplasie des Gallengangepithels (zumeist Adenokarzinom); eine Sonderstellung nimmt der Klatskin-Tumor ein, der sich im Bereich der Hepatikusgabel manifestiert. Der Tumor ist meist gut-differenziert, weist ein tubuläres bzw. papilläres Wachstum auf und hat eine ausgeprägt fibröse Komponente.
→ Epidemiologie:
→ I: Das Gallengangskarzinom tritt mit einer Inzidenz von 1/100000 Einwohnern auf und ist somit seltener als das Gallenblasenkarzinom.
→ II: Der Manifestationsgipfel liegt jenseits des 60. Lebensjahres, wobei Frauen etwas häufiger als Männer betroffen sind.
→ Ätiologie: Ursachen für die Entwicklung eines cholangiozellulären Karzinoms sind u.a.:
→ I: Primär sklerosierende Cholangitis,
→ II: Alpha-Antitrypsin-Mangel,
→ III: Colitis ulcerosa,
→ IV: Parasitäre Erkrankungen wie Leberegel,
→ V: Weitere Ursachen: Sind Anomalien der Gallengänge, intraheptische Gallensteine (z.B. Hepaticolithiasis), Leberzysten und das Caroli-Syndrom, aber auch Östrogene bzw. Progesteron und Karzinogene.
→ Klassifikationen:
→ I: Das CCC wird nach der Lokalisation unterteilt in:
→ 1) Intrahepatisches CCC: Von den intrahepatischen Gallengängen ausgehend. Diese Form wird nochmal unterteilt in eine zentrale (duktale) Form und eine periphere (häufig noduläre Form).
→ 2) Extrahepatisches CCC:
→ A) Proximales Drittel: Klatskin-Tumor, der sich im Bereich der Hepatikusgabel entwickelt.
→ B) Mittleres Drittel: Insbesondere im Bereich des Ductus choledochus (Hauptanteil).
→ C) Distales Drittel: Gallengangskarzinom im retroduodenalen Abschnitt z.B. im Bereich der Papilla vateri.
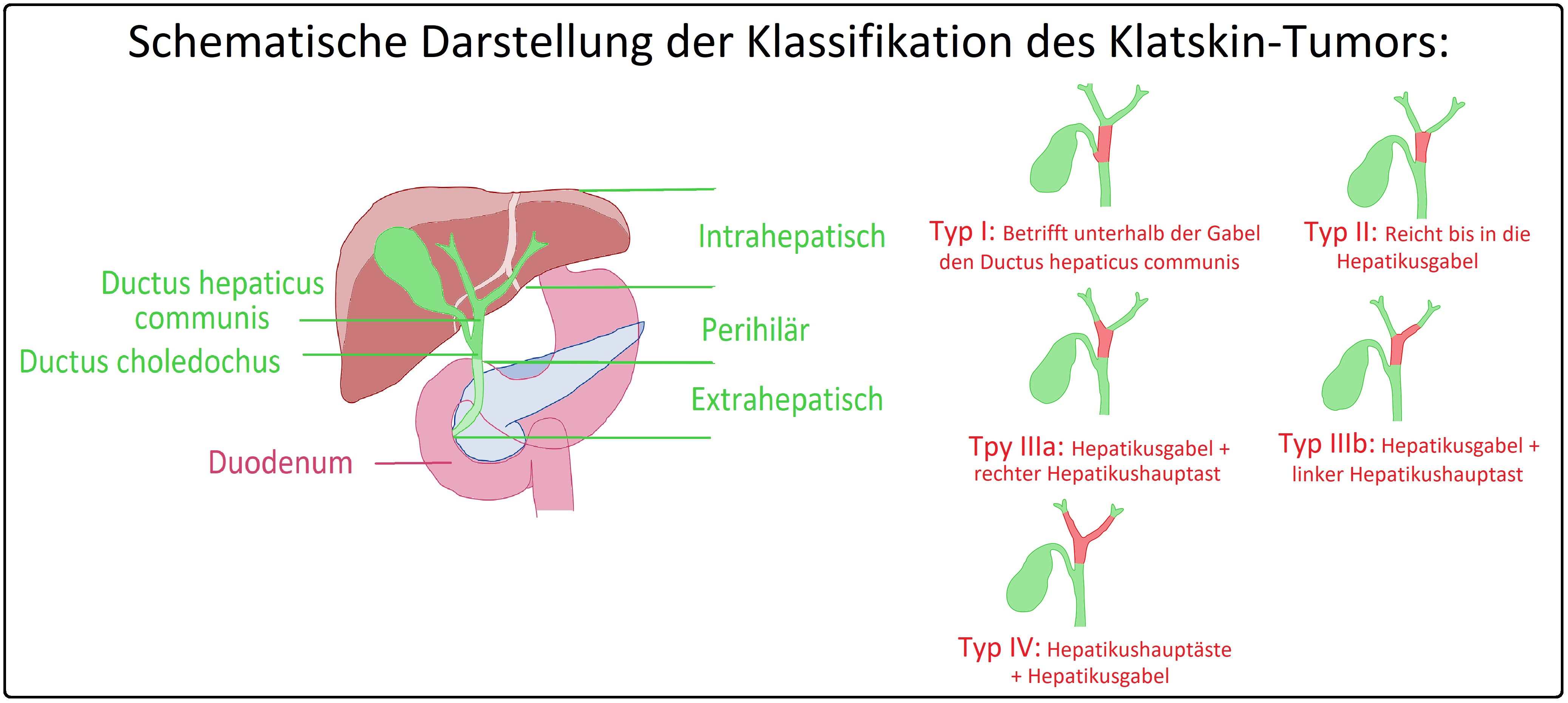
→ II: Klatskin-Tumor: Hierbei handelt es sich um ein Gallengangskarzinom im Bereich der Hepatikusgabel und wird nach in Bismuth-Corlette nochmals unterteilt in:
→ 1) Typ 1: Das Karzinom reicht bis zur Hepatikusgabel heran und betrifft den Ductus hepaticus communis.
→ 2) Typ 2: Das Karzinom bezieht die Hepatikusgabel mit ein.
→ 3) Typ 3: Das Karzinom bezieht den rechten (Typ 3a) oder den linken Hepatikushauptast (Typ 3b) mit ein.
→ 4) Typ 4: Beide Hepatikushauptäste sowie die Hepatikusgabel sind betroffen.
→ Klinisch-relevant:
→ A) Charakteristischerweise wächst der Tumor zirkulär um den Gallengang und breitet sich entlang dessen aus.
→ B) Immunhistochemisch ist das Zytokeratin CK-19 nachweisbar.
→ Klinik: Die klinische Symptomatik des cholangiozellulären Karzinoms ist häufig primär uncharakteristisch:
→ I: Klassisch schmerzloser Verschlussikterus (infolge eines langsamen Choledochus-Verschlusses und ausreichender Adaption der Gallenblasenwand an die Stauung).
→ II: Courvoisier-Zeichen: Charakterische Symptomatik bestehend aus einem Ikterus sowie einer tastbaren, schmerzlosen, prallelastischen Gallenblase.
→ III: Weitere Symptome: Sind insbesondere Gewichtsabnahme, dunkler Urin und heller Stuhl als Zeichen einer Cholestase, etc.
→ Diagnose:
→ I: Anamnese/klinische Untersuchung: Nachweis einer tastbaren, nicht schmerzhaften Gallenblase; wird als Courvoisier-Zeichen bezeichnet.
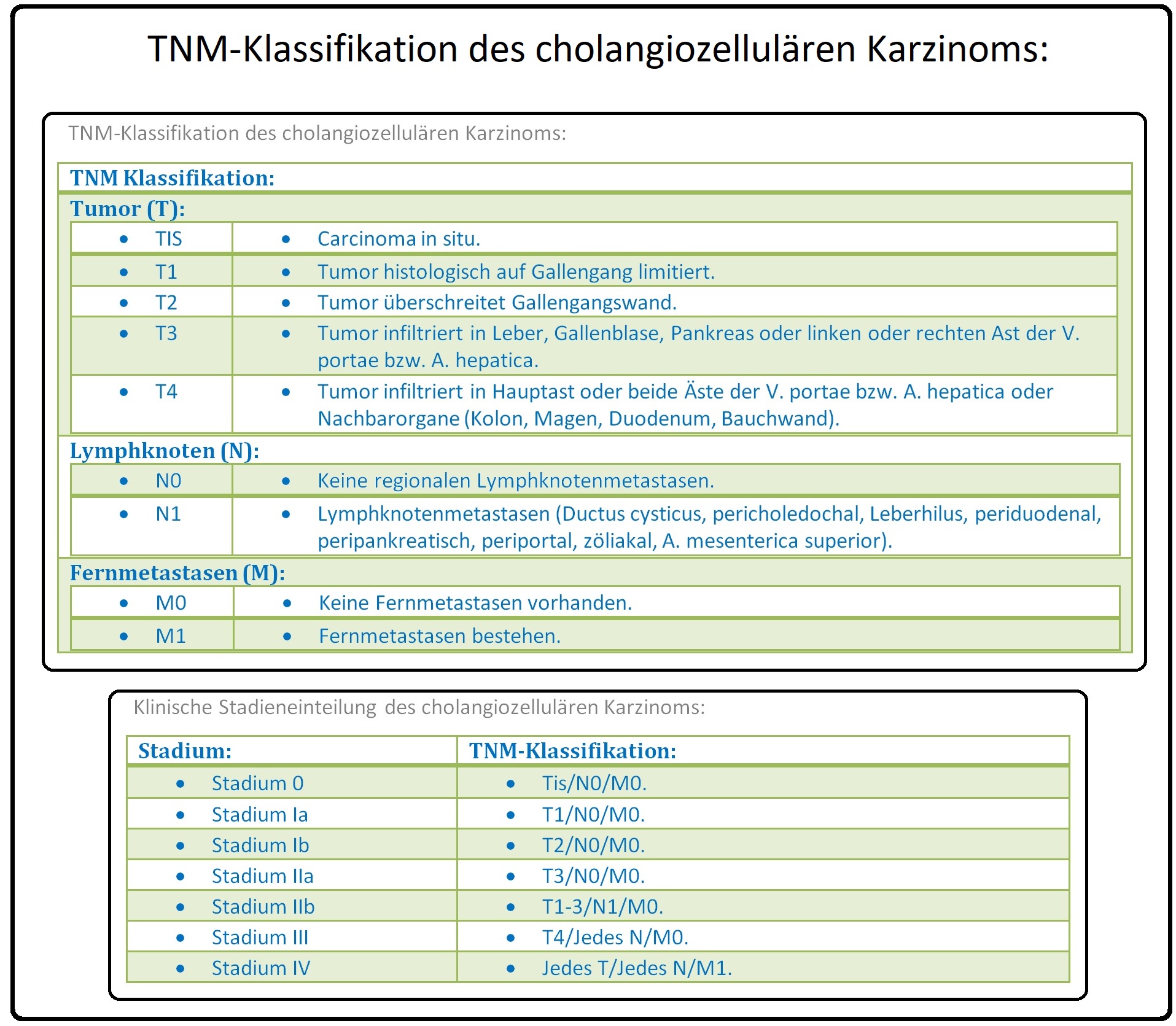
→ II: Labor: Erhöhung von CRP, BSG, der Cholestaseparameter yGT, AP, LAP und des direkten Bilirubins, sowie ein Anstieg der Tumormarker CEA (30%) und CA 19-9 (85%), CA-125 (50%).
→ III: Bildgebende Verfahren:
→ 1) Sonographie/Endosonographie: Nachweis echoarmer Strukturen, welche die Gallengänge verlegen und somit den Abfluss der Gallenflüssigkeit verhindern (erweiterte intrahepatische Gallengänge); z.T. ist ein infiltratives Wachstum nachweisbar. Weitere sonographische Befunde sind u.a. regionale Metastasierung (z.B. Lebermetastasen) und Entwicklung eines Aszites.
→ 2) ERC/ERCP: Darstellung einer Stenose des Ductus choledochus mit gleichzeitiger Dilatation des vorgeschalteten Ganges. Evtl. Einlage einer Pigtail-Drainage zur Wiederherstellung des Gallenabflusses.
→ 3) Weitere Untersuchungsverfahren: Sind
→ A) CT/MRT: Zur Beurteilung der Tumorausdehnung und möglicher Metastasen.
→ B) MR-Angio: Hierbei lassen sich der Truncus coeliacus, insbesondere die A. hepatica und die Vena portae, sowie Gefäßinfiltrationen gut darstellen.
→ C) Weitere Untersuchungen sind Spiral-CT und Positronenemissionstomographie.
→ Differenzialdiagnose: Vom cholangiozellulären Karzinom müssen insbesondere nachfolgende gastroenterologische Erkrankungen abgegrenzt werden:
→ I: Pankreaskopfkarzinom: Auch hier bestehen die klassischen Couvoisier-Zeichen (histologische Differenzierung).
→ II: Hepatozelluläres Karzinom.
→ III: Extrahepatische Cholestase: Choledocholithiasis, Cholangitis, Gallenblasenkarzinom, Kompression der Gallenwege von außen z.B. durch Lymphknoten, Tumoren (und Lebermetastasen), Stenosen der Papilla Vateri (Papillentumoren), des Weiteren Parasiteninfektion (Askardiasis), Duodenaldivertikel etc.
→ Therapie: Die operative Therapie richtet sich nach dem Ausmaß der Tumorausbreitung.
→ I: Oberes/mittleres extrahepatisches CCC:
→ 1) Hierbei erfolgt die Entfernung der extrahepatischen Gallengänge, der Gallenblase sowie der Lymphknoten im Bereich des Lig. hepatoduodenale. Die Rekonstruktion wird mittels Hepatikojejunostomie mit konsekutiver Roux-/Y-Schlinge erreicht (= Biliodigestive Anastomose). Bei Leberbefall kann die OP durch eine Leberteilresektion erweitert werden.
→ 2) Klatskin-Tumor: Therapieoptionen stellen die Trisegmentektomie rechts (= Resektion der Leber rechts des Lig. falciforme unter Miteinbeziehung des Lobus caudatus), evtl. die Pfortaderteilresektion oder die Lebertransplantation dar.
→ II: Distales CCC: Beim distalen Gallengangskarzinom z.B. Papillenkarzinom ist eine Whipple OP (= partielle Duodenopankreatektomie + Lymphadenektomie) indiziert.
→ III: Palliative Therapie:
→ 1) Drainage: Zur Ableitung der gestauten intrahepatischen Gallenflüssigkeit bei Inoperabilität kann die Anlage eines perkutanen-transhepatischen Katheters nach außen oder nach innen mittels Pigtail-Katheter oder eine biliodigestive Anastomose (= Hepatikojejunostomie) indiziert sein.
→ 2) Afterloading: Anlage eines perkutanen Führungskatheters mit anschließender lokaler Bestrahlung (Einbringen einer Strahlungsquelle wie Iridium).
→ 3) Photodynamische Therapie: Hierbei wird ein Photosensitizer i.v. appliziert, der sich in den Tumorzellen anreichert. Anschließend wird über eine ERCP ein spezifischer Lichtleiter eingebracht, der die Photoaktivierung einleitet und konsekutiv eine Tumornekrose induziert. Alle 3-6 Monate wird das Verfahren wiederholt.
→ 4) Systemische Chemotherapie mittels z.B. Gemcitabin und Cisplatin.
→ Prognose:
→ I: Die Prognose des cholangiozellulären Karzinoms ist im Allgemeinen sehr ungünstig.
→ II: Gerade die Klatskin-Tumoren sind zumeist zum Zeitpunkt der Diagnosestellung in den Ductus hepaticus, die Vena portae und die A. hepatica, evtl. auch ins umliegende Leberparenchym infiltriert (inoperabel), sodass die 5-Jahresüberlebenschance bei diesen Patienten sehr gering ist.
→ III: Demgegenüber haben weiter distal gelegene Tumoren eine bessere Prognose.
→ IV: Bei nicht-resektablen Patienten beträgt das mittlere Überleben 6-12 Monate.