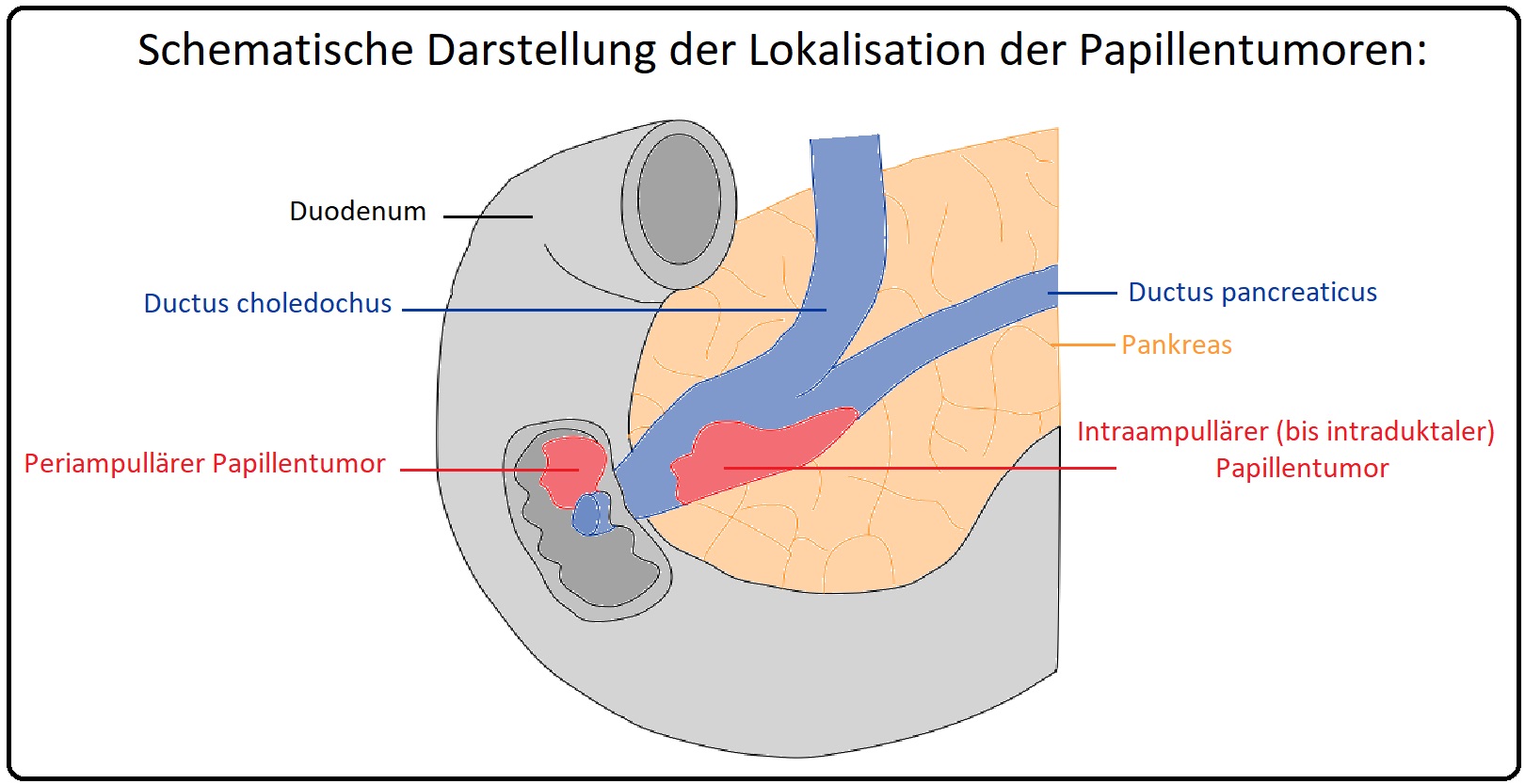
→ Definition: Unter die selten auftretenden Papillentumoren werden sowohl die benignen adenomatösen Raumforderungen, als auch die malignen Neoplasien im Bereich der Papilla Vateri (periampullär oder intraampullär) zusammengefasst.
→ Epidemiologie:
→ I: Die Papillentumoren sind insgesamt sehr selten; die Häufigkeit für benigne Tumoren wird mit ca. 0,04-0,62 %, für Papillenkarzinome mit 0,063-0,21% angegeben, wobei insbesondere Männer nach dem 65. Lebensjahr betroffen sind.
→ II: Eine gehäufte Prävalenz für Papillenadenome bzw. -karzinome findet man bei Patienten mit familiärer adenomatöser Polyposis; hierbei stellt die Papillenregion eine Prädilektionsstelle dar.
→ Klinisch-relevant: Patienten mit Polyposis-Syndromen (z.B. familiäre Adenomatosis coli, Peutz-Jeghers-Syndrom, Cowden-Syndrom) haben ein 100-fach erhöhtes Risiko für die Entstehung eines ampullären Karzinoms.
→ Pathogenese: Wie beim kolorektalen Karzinom hat sich auch bei den Papillentumoren das Vorliegen einer Adenom-Karzinom-Sequenz etabliert.
→ I: Bei den Papillenkarzinomen handelt es sich überwiegend um Adenokarzinom, die makroskopisch in lobulierte und infiltrierende Raumforderungen unterteilt werden.
→ II: Des Weiteren existierte eine Klassifikation der Papillenneoplasie in polypoider -, ulzerativer und gemischter Typ. Der polypoide Typ kann wiederum in ein exophytisches und intramurales Wachstum klassifiziert werden.
→ III: Histologie: Auch histologisch gibt es eine Unterteilung in:
→ 1) Intestinaler Typ: Mit siebförmig imponierenden Drüsen und
→ 2) Pankreatobiliärer Typ: Der ähnliche bzw. gleiche Eigenschaften wie das duktale Adenokarzinom des Pankreas aufweist.

→ Klinik: Papillentumoren können aufgrund ihrer Lokalisation in der Ampulle in frühen Stadien klinische Symptome hervorrufen, jedoch existieren keine verlässlichen Frühsymptome:
→ I: Abdominelle, z.T. kolikartige Schmerzen, Übelkeit und Erbrechen.
→ II: Cholestase, Verschlussikterus, evtl. auch Pruritus.
→ III: Des Weiteren können Anämie (nicht selten aufgrund von okkulten Darmblutungen), rezidivierende Cholangitiden und Pankreatitiden auftreten.
→ IV: Bei malignen Prozessen kann sich die typische B-Symptomatik mit:
→ 1) Müdigkeit und Leistungsminderung.
→ 2) Appetitlosigkeit und Gewichtsverlust sowie
→ 3) Nachtschweiß aufzeigen.
→ Diagnose:
→ I: Anamnese/klinische Untersuchung:
→ 1) In der Anamnese Nachweis von rezidiverende Cholangitiden, Pankreatitiden oder einer familiär- adenomatöse Polyposis.
→ 2) Bei der klinischen Untersuchung möglicher Nachweis eines intermittierenden schmerzlosen Ikterus und evtl. Courvoisier-Zeichen als schmerzlose palpable Gallenblase.
→ II: Labor: Mit Zeichen einer Anämie, Cholestase (yGT, AP, Bilirubin) sowie Bestimmung der Leberenzyme mit GOT und GPT. Bei Pankreatitis bzw. Cholangitis Nachweis der Entzündungsparameter (Leukozytose Erhöhung der BSG und CRP) sowie der Pankreasenzyme (Amylase und Lipase). Bei malignen Neoplasien können zur Verlaufskontrolle die Tumormarker CEA und CA19-9 herangezogen werden.
→ III: Bildgebung:
→ 1) Oberbauchsonographie: Möglicher Nachweis eines dilatierten Ductus choledochus (extrahepatische Cholestase) oder der Pankreasgänge häufig mit Gallenblasenhydrops. Insbesondere bei kleineren Tumoren (< 2cm) ist die Diagnose schwierig. Jedoch ist ein Gangabbruch ist immer tumorverdächtig, auch wenn sich sonographisch keine Raumforderung abgrenzen lässt. Zudem können mögliche Metastasen oder Lymphknotenvergrößerungen ausgeschlossen werden.
→ 2) Die Diagnose des Papillentumors wird mittels Ösophagogastroduodenoskopie oder ERCP mit Biopsie gestellt. Endoskopisch zeigt sich nicht selten eine vergrößerte Papille Vateri; Ulzerationen, Indurationen sowie eine submuköse oder intraduktale Infiltration ist immer ein Hinweis für Malignität. Gesichert wird die Diagnose (mindestens 6 Biopsien) histologisch.
→ 3) Zum weiteren Staging mit Beurteilung der Tumorgröße, lokalen Infiltration und möglichen Lymphknotendiagnostik hat sich der endoskopische Ultraschall (EUS) oder ggf. der intraduktale Ultraschall (IDUS) durchgesetzt.
→ Differenzialdiagnose: Von den Papillentumoren müssen insbesondere nachfolgende Erkrankungen abgegrenzt werden:
→ I: Cholestase anderer Genese.
→ II: Gallengangsstenose aufgrund einer chronischen Pankreatitis.
→ III: Pankreaskopfkarzinom und nicht zuletzt
→ IV: Das cholangiozelluläres Karzinom, etc.
→ Therapie: Vorherrschende Therapieziele sind die Beseitigung des Abflusshindernisses sowie die vollständige Abtragung des Papillentumors, um eine histologische Auswertung zu erlangen und das Rezidivrisiko zu senken.
→ I: Bei benignen Tumoren ist eine transduodenale endoskopische Papillenexzision indiziert; dies ist auch bei größeren, lateral ausgedehnten beningen Tumoren sowie längerstreckigem intraduktalem Wachstum möglich.
→ II: Bei invasiven Karzinomen ist die Therapie der ersten Wahl die partielle Pankreatikoduodenektomie nach Whipple. Postoperative Komplikationen sind Abszess- und Fistelbildungen, Magenentleerungsstörungen sowie Nachblutungen (40% der Fälle).
→ III: Palliative Interventionen: Umfassen insbesondere:
→ 1) Chemotherapie bei metastasierten periampullären Karzinomen (analog dem Pankreaskarzinom).
→ 2) Endoskopische Platzierung einer Kunststoffprothese oder eines Metall-Stents wird bei einer Lebenserwartung über 3 Monate empfohlen.
→ 3) Chirurgische Anlage einer biliodigestiven Anastomose zur Drainage der Galle.
→ IV: Postoperative Komplikationen: Hierzu zählen u.a.:
→ 1) Nach endoskopisch Resektion können Pankreatitiden, Blutungen, Perforationen und selten auch Gallengangsstrikturen, etc. nachgewiesen werden.
→ 2) Rezidive: Eine relativ häufige Komplikation ist die Manifestation von Rezidiven benigner Tumoren nach endoskopischer Resektion, die wiederum reseziert werden können.
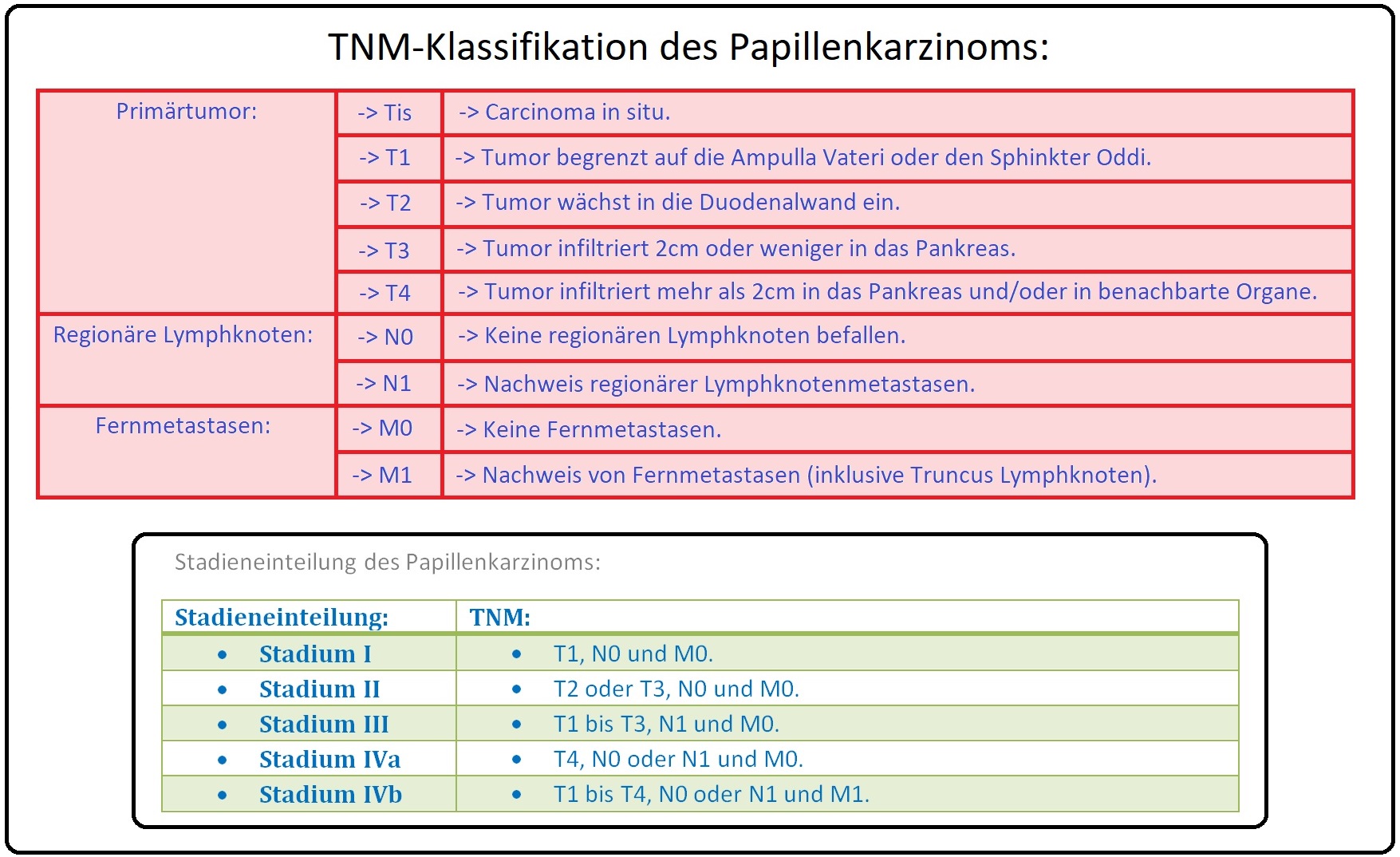
→ Prognose: Der Erkrankungsverlauf und die Prognose sind insbesondere vom Stadium und der Größe des Papillentumors abhängig und aufgrund der frühzeitigen symptomatischen Manifestation wird es nicht selten im Anfangsstadium diagnostiziert:
→ I: So sind auch die Papillenkarzinome zur Diagnosestellung in bis zu 80% operabel.
→ II: Die 5-Jahresüberlebensrate liegt bei 45-65% und ist vor allem vom Lymphknotenstatus und einer evtl. Infiltration in das Pankreas abhängig.
→ III: Präventive Maßnahmen umfassen u.a.:
→ 1) Die regelmäßige endoskopische Kontrolle (alle 3-5 Jahre bei Fehlen von Adenomnachweisen) von Patienten mit familiärer adenomatöser Polyposis ab dem 30. Lebensjahr.
→ 2) Nach vollständiger endoskopischer oder chirurgischer Resektion sind auch hier regelmäßige Kontrolluntersuchungen nach 3, 6 und 12 Monaten und anschließend jährlich über mindestens 3 Jahre obligat (hierdurch können Rezidive frühzeitig erkannt und therapiert werden).