→ Definition: Bei der primär biliären Zirrhose handelt es sich um eine autoimmunologisch bedingte, chronisch-nichteitrige Cholangitis mit fortschreitender Destruktion der kleinen Gallengänge sowie Gallengangsproliferation. Das Endstadium ist durch eine biliäre Fibrose und Leberzirrhose charakterisiert.
→ Epidemiologie:
→ I: Die Inzidenz für die pimäre biliäre Zirrhose variiert geographisch deutlich und liegt in den USA und Deutschland zwischen 3-4/100000 Einwohnern (am häufigsten in Nordeuropa).
→ II: Frauen sind 10 mal häufiger als Männer betroffen, wobei sich die klinische Symptomatik insbesondere zwischen dem 40.-60. Lebensjahr manfestiert.
→ III: Ein erhöhtes Erkrankungsrisiko besteht v.a. bei Verwandten 1. Grades sowie eineiigen Zwillingen (Konkordanzrate > 60%).
→ Ätiopathogenese:
→ I: Im Mittelpunkt der Pathogenese steht die Destruktion und der Verlust von Gallengangsepithel in Assoziation mit Infiltraten, bestehend aus eosinophilen Granulozyten (v.a. in der Frühphase der Erkrankung), T-Lymphozyten, CD8+-zytotoxischen und nicht zuletzt CD4+-T-Helferzellen. Ursache der Autoimmunreaktion ist die Präsentation von Autoantigenen auf der Oberfläche des Gallengangsepithels, die konsekutiv eine zelluläre bzw. humorale Immunreaktion hervorruft. In der Folge kommt es zu einer vermehrten CD4+-T-Helferzellen vermittelten Sezernierung von Zytokinen, die über CD8+-zytotoxische-T-Lymphozyten zu einer Schädigung bzw. Verlustes durch Nekrose und Apoptose des Gallengangsepithels führt.
→ II: Antimitochondriale Antikörper: (= AMA) Hierbei handelt es sich um Autoantikörper, die sich gegen mitochondriale Proteine (als Autoantigene) richten und bei der primär biliäre Zirrhose eine hohe Sensitivität aufweisen. Je nach Art der Autoantigene und der Häufigkeit werden verschiedene AMA-Typen unterschieden.
→ III: Autoantigene:
→ 1) Autoantigene sind bei der PBC Komponenten eines Multienzymkomplexes, der in den Mitochondrien lokalisiert ist und die oxidative Decarboxylierung der Alpha-Oxosäure einleitet. Am häufigsten zeigen sich Autoantikörper gegen die Komponente E2 (E2-Untereinheit des Pyruvatdehydrogenasekomplexes = PDC-E2) des Multienzymkomplexes.
→ 2) Des Weiteren existieren bei der PBC Autoantikörper wie Anti-gp-210, Anti-sp 100, Anticyclin A, Antinucleopurin P62, etc. gegen Proteine der Kernmembran; sie sind weniger häufig, jedoch sehr spezifisch für die primär biliäre Zirrhose.
→ IV: Umweltfaktoren: Es gibt Bakterien wie z.B. Escherichia coli oder das gram-negative Novosphinobium aromaticivorans, die potenzielle Träger eines kreuzreagierenden Epitops (Lipoyldomäne) sind, das eine ausgeprägte Homologie zu der o.g. E2-Untereinheit hat.
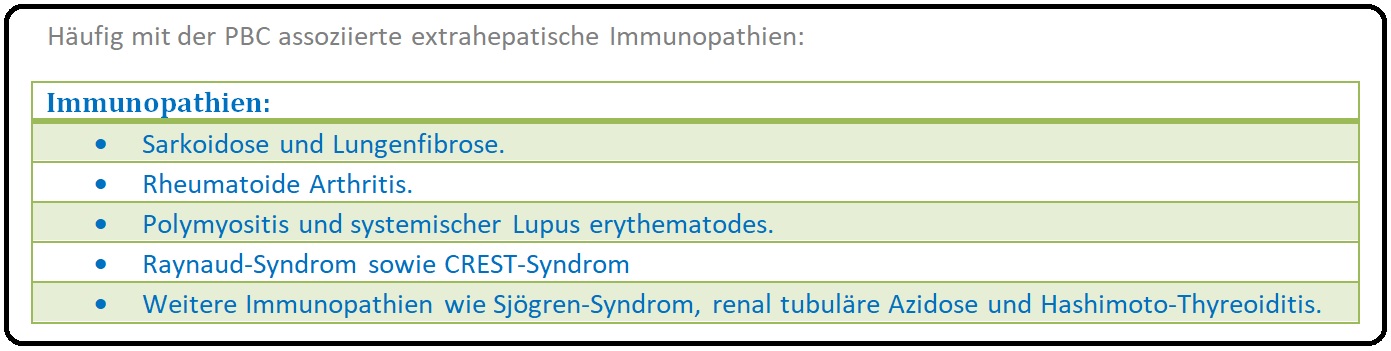
→ Klinik: Die Erkrankung bleibt häufig über einen längeren Zeitraum (im Mittel über 6 Jahre) klinisch stumm. Evtl. können in dieser Phase begleitende Autoimmunerkrankungen wie die Hashimoto-Thyreoiditis, Sarkoidose, rheumatoide Arthritis, das Sicca-Syndrom vorhanden sein. Typische klinische Symptome der primär biliären Zirrhose sind insbesondere:
→ I: Müdigkeit und Abgeschlagenheit.
→ II: Generalisierter quälender und therapierefraktärer Pruritus noch vor Auftreten des Ikterus.
→ III: Gastrointestinal:
→ 1) Uncharakteristische epigastrische Beschwerden sowie Hepatomegalie.
→ 2) Maldigestion mit Steatorrhö durch reduzierte Gallensäureexkretion.
→ IV: Osteomalazie durch Vitamin-D-Resorptionsstörung.
→ V: Hyperlipidämie, selten Xanthome und Xanthelasmen (initial im Bereich der Ober und Unterlider, später auch den Gelenken und Sehnen sowie den Handinnenflächen und am Gesäß).
→ IV: Im Spätstadium manifestieren sich u.a.:
→ 1) Portale Hypertension,
→ 2) Aszites bis hin zur
→ 3) Hepatischen Enzephalopathie (als Zeichen einer Dekompensation).
→ Diagnose:
→ I: In der Anamnese bzw. klinischen Untersuchung Exploration möglicher assoziierter Immunopathien sowie der charakteristischen klinischen Symptomatik (Hyperpigmentierung, Juckreiz, Hepatomegalie).
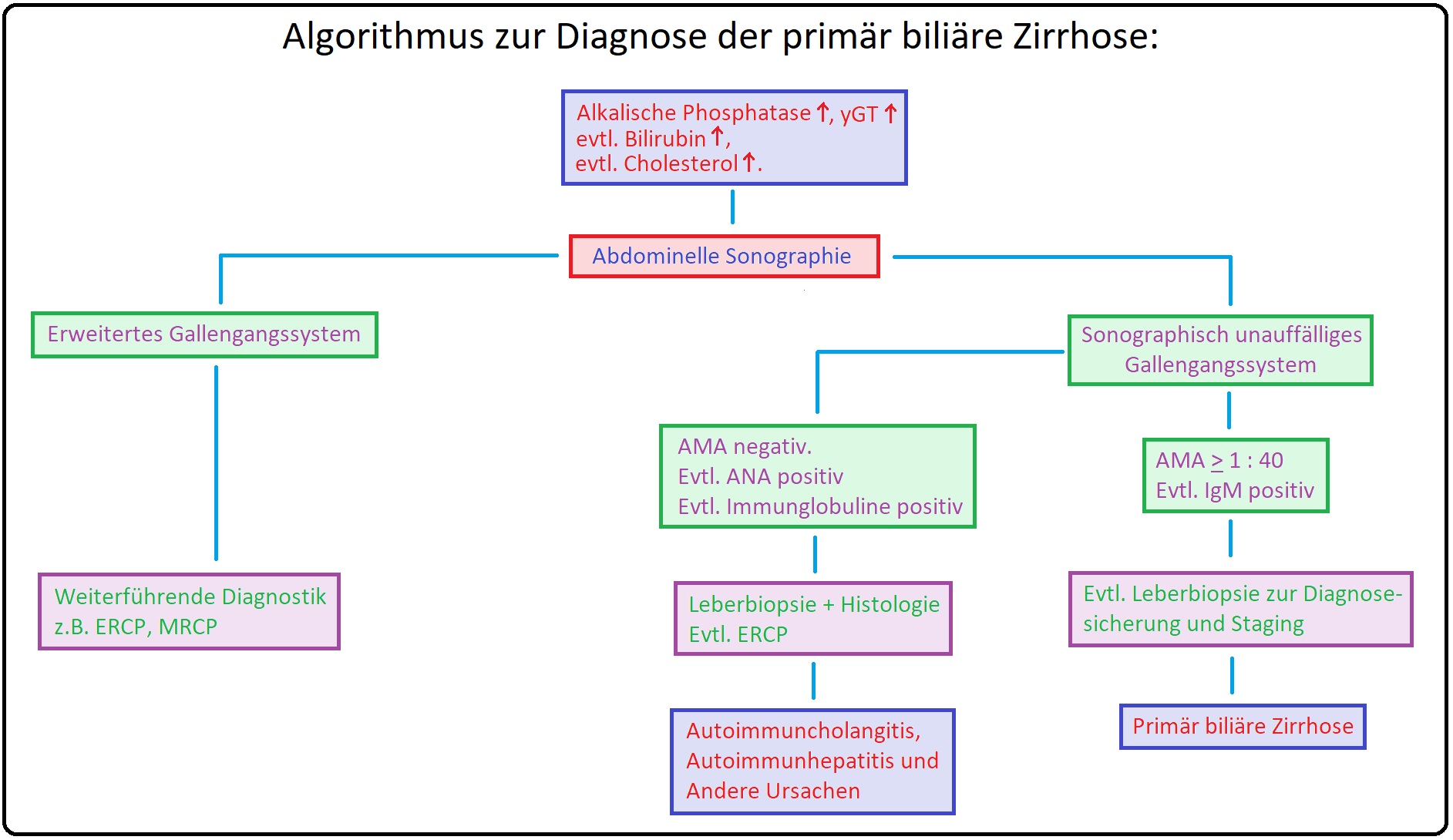
→ II: Labor:
→ 1) Die Erhöhung der alkalischen Phosphatase und yGT als Cholestaseparameter sind frühes und obligates Symptom der PBC.
→ 2) Der Anstieg des Bilirubins spricht für ein fortgeschrittenes Stadium und korreliert direkt mit der Prognose (Bilirubin 34µmol/l mittleres Überleben bei 4 Jahren).
→ 3) Antimitochondriale Antikörper: (Nachweis z.B. mittels indirekter Immunfluoreszens) Niedrige AMA-Titer sind sehr unspezifisch, währenddessen hohe Titer (95% der Fälle) eine ausgeprägte Sensitivität und Spezifität für die Diagnose der primär biliären Zirrhose aufweisen.
→ 4) Antinukleäre Antikörper: Können insbesondere bei AMA-negativen Patienten mit einer typischen PBC-Befundkonstellation nachgewiesen werden; dies kann für ein Overlap-Syndrom zwischen PBC und Autoimmunhepatitis sprechen (ist jedoch nicht ein verlässlicher Marker).
→ 5) Bei zweifelhaften Fälle sollte eine Bestimmung der Immunglobulinfraktionen erfolgen, die typischerweise mit einer IgM-Erhöhung einhergeht.
→ III: Bildgebung:
→ 1) Sonographie/CT: Weisen keine spezifische Bildmorphologie auf, vielmehr dienen diese Untersuchungsverfahren dem Ausschluss von intra- und extrahepatischen Gallensteinen, Neoplasien im Bereich von Leber, ableitenden Gallengängen und Pankreas, etc.
→ 2) ERCP: Hier zeigen sich Kaliberunregelmäßigkeiten, abweichende Verläufe sowie Rarefizierung der intrahepatischen Gallengänge.
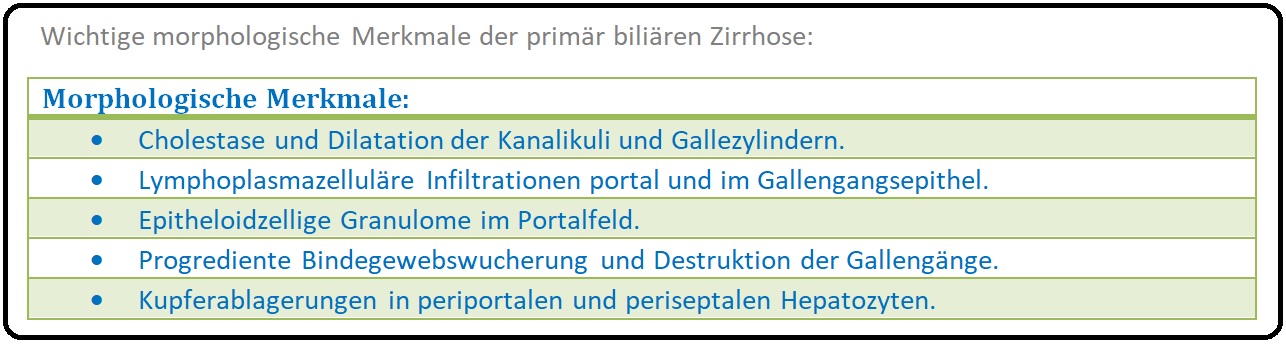
→ 3) Leberbiopsie: Mit Histologie stellt die definitive Sicherung der primär biliären Zirrhose dar:

→ Klinisch-relevant: Die Diagnose einer PBC kann gestellt werden, wenn AP und yGT über einen Zeitraum von 6 Monaten erhöht sind, AMA mit einem Titer von > 1:40 vorliegt und der histopathologische Befund die typischen Charakteristka aufweist.
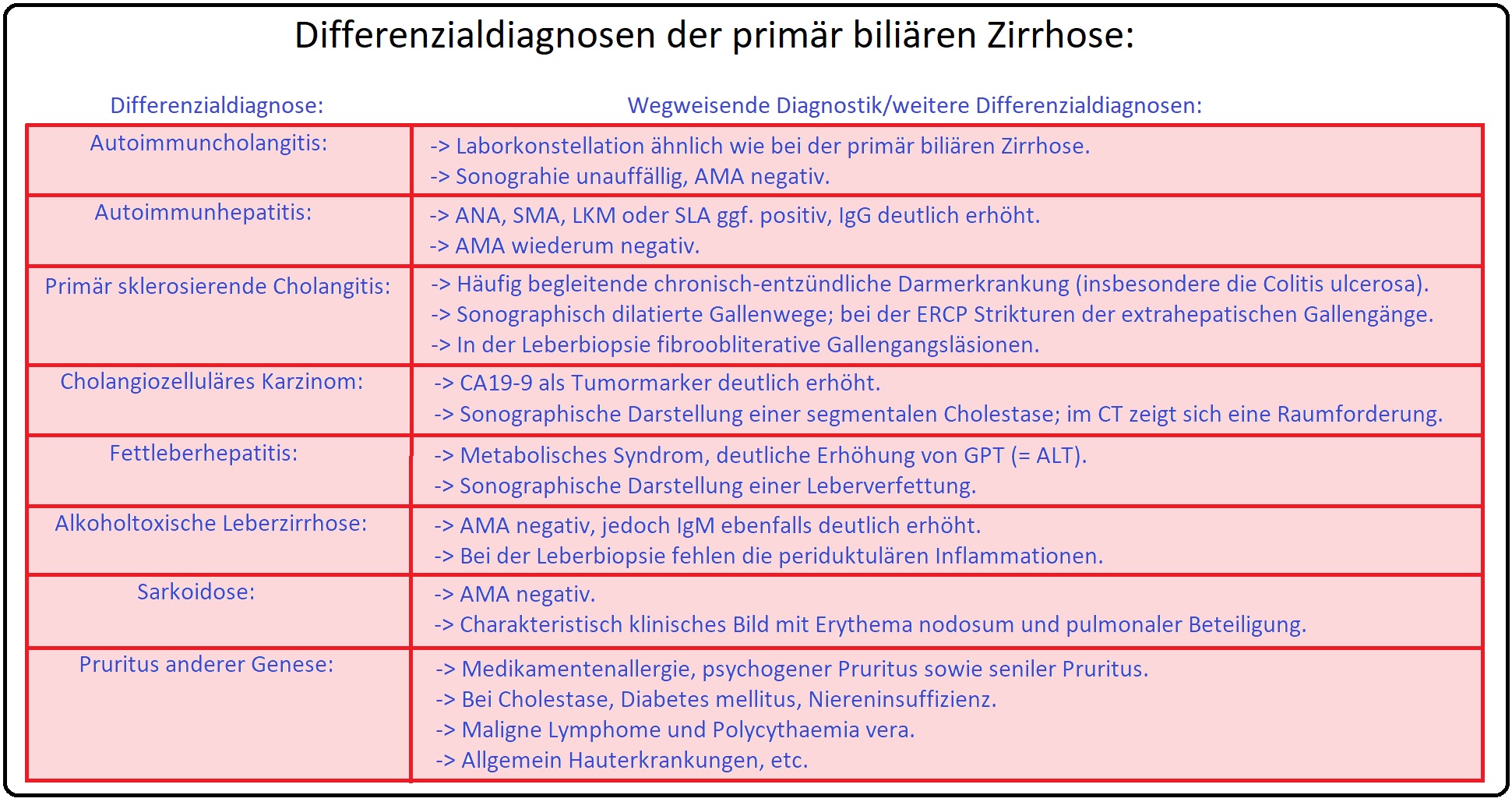
→ Differenzialdiagnose: Von primär biliäre Zirrhose müssen insbesondere nachfolgende Erkrankungen abgegrenzt werden:
→ I: Autoimmuncholangitis sowie primär sklerosierende Cholangitis und cholangiozelluläres Karzinom.
→ II: Fettleberhepatitis, und alkoholtoxische Leberzirrhose, aber auch cholestatisch verlaufende Virushepatitis sowie medikamententoxische Hepatitis.
→ III: Sarkoidose.
→ Therapie: Eine kausale Therapie für die primär billiäre Zirrhose.
→ I: Medikamentöse Therapie: Die Ursodesoxycholsäure (= UDCA)ist heutzutage das Medikament der 1. Wahl bei der Behandlung der PBC und sollte möglichst frühzeitig in einer Dosis von 15mg/kgKG/d appliziert werden. Die Wirkungsmechanismen bei UDCA bei Cholestase sind u.a.:
→ 1) Protektion von Cholangiozyten gegen toxische Wirkung von Gallensäuren.
→ 2) Stimulation der Gallensekretion.
→ 3) Entgiftung von hydrophoben Gallensäuren und nicht zuletzt
→ 4) Hemmung der Apoptose und Nekrose von Hepatozyten.
Unter Ursodesoxycholsäure verbessern sich nicht nur die Laborbefunde (klinisch-chemischen Befunde sowie Entzündungsparameter) sondern auch langfristig die Leberhistologie. Bei fehlender Besserung kann UDCA mit Budenosid in frühen Stadien kombiniert werden. Prednisolon (in Kombination mit UDCA) hat sich in einer Dosierung von 10-15mg/d insbesondere in der Behandlung des Overlap-Syndrom von PBC und Autoimmunhepatitis mit erhöhter Aminotransferase und IgG-Fraktion etabliert.
→ II: Symptomatische Therapie:
→ 1) Der Juckreiz kann mit Ingelan-Puder behandelt werden; weitere Substanzen sind Colestyramin oder Cholestabyl (hemmen die Gallensäurenrückresorption) sowie Naltrexon haben einen positiven Effekt auf den Pruritus.
→ 2) Besteht ein Malabsorptionssyndrom ist die Gabe der fettlöslichen Vitamine (A, D, E, K) indiziert.
→ 3) Bei Steatorrhoe sind mittelkettige Triglyzeride obligat.
→ III: Ultima ratio stellt die Lebertransplantation dar und sollte insbesondere bei steigenden Bilirubinwerten erwogen werden.
→ Prognose: Eine Spontanremission existiert bei der primär biliären Zirrhose nicht und die durchschnittliche Lebenserwartung bei symptomatischen Patienten beträgt in der Mitte 12 Jahre. Vor allem das histopathologische Stadium und die Bilirubinwerte sind von prognostischer Bedeutung. Bei einem erhöhter Bilirubinwert > 6mg/dl liegt die mittlere Überlebenszeit meist nur noch unter 2 Jahren.