→ Definition: Wird auch als portosystemische Enzephalopathie (= PSE) bezeichnet. Es handelt sich um eine Komplikation des Leberzerfallskomas bzw. Leberausfallkomas mit vermehrter Retention neurotoxischer Substanzen (insbesondere Ammoniak, aber auch Phenole, Mercaptane etc.), Störungen der Astrozytenfunktion, Dysfunktion der Blut-Hirn-Schranke sowie Störungen der Aminosäurebalance (verzweigkettig-aromatisch). Klinisches Korrelat sind psychopathologische und extrapyramidale Symptome, die im weiteren Krankheitsverlauf zu Vigilanzstörungen und Koma führen.
→ Klassifikation:
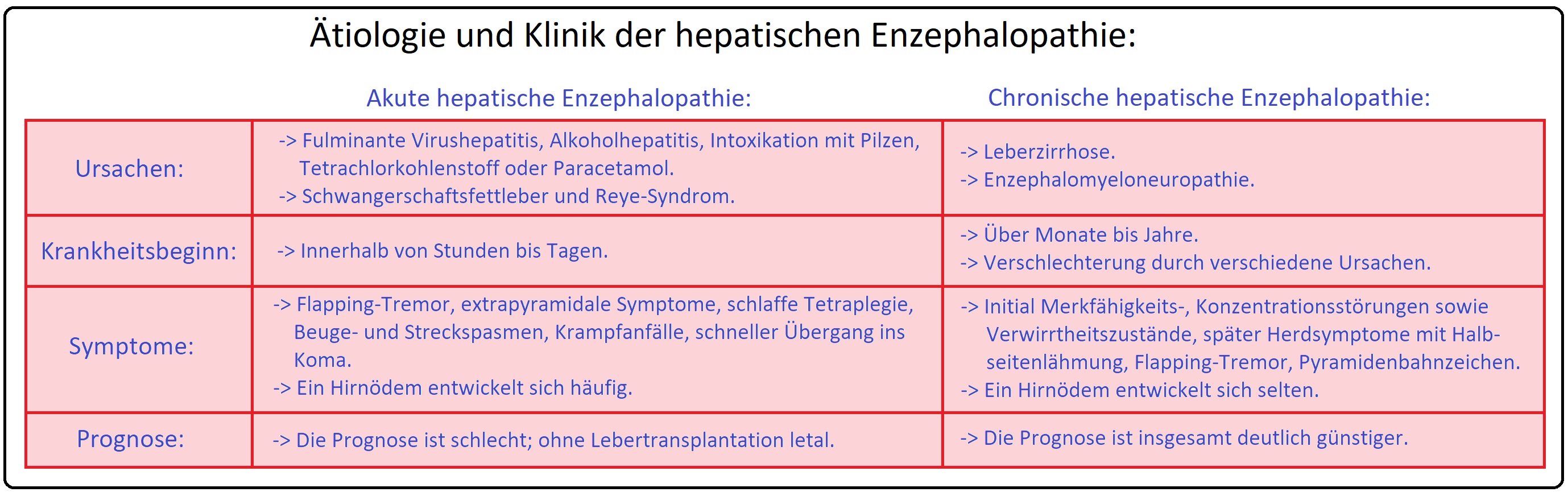
→ I: Leberzerfallskoma: Ist ein akuter Ausfall der Leber als Folge einer massiven Leberzellnekrose, hervorgerufen durch:
→ 1) Virushepatitiden: Hepatitis D, Hepatitis B, Hepatitis A und Hepatitis C.
→ 2) Medikamente wie Halothan, Paracetamol, MAO-Hemmer, Valproat, Dilsulfiram, Cotrimoxazol, NSAR wie Pirprofen etc.
→ 3) Amantadin, Tetrachlorkohlenstoff,
→ 4) Massive hepatische Infiltrationen im Zuge einer akuten Leukämie, Morbus Hodgkin etc.
→ 5) Andere Ursachen sind das Budd-Chiari-Syndrom, Morbus Wilson, portosystemische Shuntoperation, primär biliäre Zirrhose, etc.
→ II: Leberausfallkoma: Dieses verläuft langsamer als das Leberzerfallskoma und ist Folge einer Leberzirrhose (latenter Verlauf). Provoziert wird es durch Alkoholexzesse, Diätfehler, Varizenblutungen, Elektrolytverschiebungen (Hypokaliämie), etc.
→ Pathogenese: Aufgrund einer Leberzirrhose unterschiedlicher Genese.
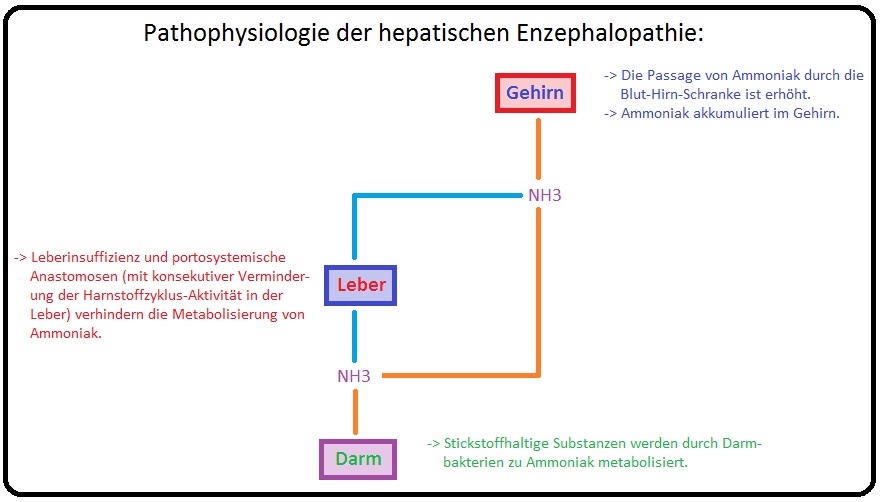
→ I: Fehlende Entgiftung der Leber von neurotoxischen Stoffen wie Ammoniak, Mercaptane, Phenolderivate sowie von kurz- und mittelkettige Fettsäuren. Infolge des Umgehungskreislaufes der Leber gelangt das durch die Zersetzung von Eiweißen und Fetten gewonnene Ammoniak in den großen Kreislauf und wird nicht in der Leber metabolisiert. Ammoniak akkumuliert im Gehirn und bewirkt über eine Steigerung der Glutaminsynthese in den Astrozyten eine Anschwellung dieser. Folge ist die Entwicklung eines akuten Hirnödems. Weitere pathologische Veränderungen sind u.a.:

→ II: Leberinsuffizienz mit verminderter Syntheseleistung (Cholinesterase, Albumin, Gerinnungsfaktoren 1972 = II, VII, IX, X).
→ III: Ausbildung von Kollateralkreisläufen mit fehlender First-pass-Clearance der Leber.
→ IV: Provozierende Faktoren: Orale Proteinzufuhr, bakterielle Infektionen, Varizenblutungen, chirurgische Eingriffe, forcierte Diurese, Aszites-Punktion, Elektrolytverschiebungen (Hypokaliämie), Störungen des Säure-Base-Haushaltes. So verursacht eine metabolische Alkalose eine verminderte Bikarbonatausscheidung mit konsekutiv vermehrter Aufnahme von Ammoniak im Gehirn.
→ Klinisch-relevant: Faktoren für eine Verschlechterung einer PSE:
→ A) Vermehrte Ammoniakbildung im Darm:
→ 1) Bei GIT-Blutungen (1000ml = 200g Eiweiß),
→ 2) Aber auch bei eiweißreichem Essen und
→ 3) Obstipation.
→ B) Vermehrte Ammoniakdiffusion ins Gehirn durch Alkalose.
→ C) Vermehrter Eiweißkatabolismus bei Infekten
→ C) Iatrogen: Durch Gabe von Benzodiazepine, Sedativa, Analgetika und durch intensive Diuretika-Therapie.
→ Klinik: Bevor sich das Beschwerdebild im Sinne der klinischen Stadieneinteilung der hepatischen Enzephalopathie manifestiert, leiden die meisten Patienten bereits an kognitiven Störungen wie z.B. Aufmerksamkeitsdefiziten, Störungen in der Geschwindigkeit der Informationsverarbeitung und Psychomotorik, Affektlabilität, depressiven Verstimmungen, Kopfschmerzen etc.:

→ I: Stadium 0: Asymptomatische HE, jedoch pathologische psychometrische Tests.
→ II: Stadium 1: Beginnende Schläfrigkeit, Stimmungsschwankungen, Konzentrationsstörungen, Verlangsamung, leichtgradige Verwirrung, verwaschene Sprache.
→ III: Stadium 2: Zunehmende Schläfrigkeit, Apathie, Veränderung der Schriftprobe und des EEG´s, Asterixis = grobschlägiges Händezittern (flapping Tremor)
→ IV: Stadium 3: Patient schläft stets, ist jedoch erweckbar, nachweisbarer Korneal- und Sehnenreflex, einsetzender foetor hepaticum. Der flapping Tremor ist noch vorhanden, EEG verändert.
→ V: Stadium 4: Leberausfallkoma: Tiefer Schlaf, durch Schmerzreiz nicht mehr erweckbar, Kornealreflex negativ, Foetor hepaticus, Flapping tremor negativ, EEG Veränderungen sind vorhanden.
→ Komplikationen: Typisches Komplikationen sind:
→ I: Disseminierte intravasale Koagulopathie (= DIC),
→ II: Gastrointestinale Blutungen, meist im Bereich des Magens und
→ III: Hirnödem.
→ Diagnose:
→ I: Anamnese/Klinische Untersuchung:
→ 1) Pathologischer Reitan-Test: Hierbei handelt es sich um einen Rechentest, bei dem die Zahlen numerisch angeordnet werden müssen.
→ 2) Pathologische Schriftprobe (mit u.a. irregulärem Wort- und Zeilenabstand).
→ 3) Frühzeitiger Nachweis von gesteigerten Eigenreflexen; insbesondere bei fulminanter Leberinsuffizienz mit konsekutiv zunehmenden intrakraniellen Druck manifestiert sich ein Einklemmungssyndrom mit Muskeltonuserhöhung und Streckkrämpfen.
→ II: Labor: Es existieren keine spezifischen Laborparameter.
→ 1) Natrium (durch Vasodilatation und gesteigerte ADH-Produktion) und Kalium (= Hypokaliämie durch rezidivierendes Erbrechen, Diarrhoe, sekundären Hyperaldosteronismus) erniedrigt.
→ 2) Bilirubin erhöht (als Ausdruck der Lebeschädigung).
→ 3) Albumin, Quick, Cholinesterase und AT-III sind Aussruck der verminderten hepatischen Syntheseleistung.
→ 4) Ein Abfall der Gerinnungsfaktoren, beginnend mit Faktor VII, spricht eher für einen ungünstigeren Krankheitsverlauf.
→ 5) Bei akuter hepatischer Schädigung zeigt sich ein starker Transaminase-Anstieg als Zeichen einer Leberzellnekrose; ein Transaminaseabsturz spricht für eine zerstörte Leber.
→ 6) Ammoniak > 100µg/dl (Blutentnahme aus einer ungestauten Vene) bestätigt die Diagnose und korreliert mit der Schwere der neurologischen Symptome.
→ III: EEG: Im Elektroenzephalogramm zeigt sich in Abhängigkeit vom Schweregrad der hepatischen Enzephalopathie initial eine intermittierende, später eine kontinuierliche Verlangsamung des Grundrhythmus. Charakteristisch sind triphasische Wellen und eine generalisierte Delta-Aktivität.
→ Differenzialdiagnose: Von der hepatischen Enzephalopathie müssen insbesondere nachfolgende Erkrankungen abgegrenzt werden:
→ I: Das chronische Subduralhämatom, das infolge einer erhöhten Blutungsneigung bei schwerer Leberinsuffizienz entstehen kann und häufig mit psychopathologischen Symptomen einhergeht.
→ II: Akute Porphyrie: (z.B. akut intermittierende Porphyrie, etc.) Charakteristika sind psychotische Symptome, Vigilanzstörungen sowie eine symmetrische vorwiegend motorische Polyneuropathie.
→ III: Bei der Hyperthyreose existieren u.a. ein fleinschlägiger hochfrequenter Tremor, evtl. auch choreoathetotische Hyperkinesien zusammen mit Schlafstörungen und Affektlabilität.
→ IV: Alkoholinduzierte Gehirnschäden, aber auch Alkoholentzugssyndrom und die Wernicke-Enzephalopathie.
→ V: Enzephalopathie anderer Genese z.B. die urämische Enzephalopathie im Rahmen einer dekompensierten Niereninsuffizienz oder eines akuten Nierenversagens.
→ VI: Weitere Differenzialdiagnosen sind u.a. Meningitis und Enzephalitis, etc.
→ Therapie:
→ I: Allgemeinmaßnahmen:
→ 1) Kontrolle von RR, Puls, Atmung und Temperatur.
→ 2) Gewichtskontrolle, Ein- und Ausfuhrkontrolle.
→ 3) Adäquate parenterale, kalorische (2000kcal/d), proteinarme Ernährung (Magensonde: 0,5g Proteine/kgKG/d): Dies senkt den EW-Metabolismus (Merkaptane, Ammoniak) durch Verminderung des EW-Katabolismus.
→ Klinisch-relevant: Nach Varizenblutungen oder bei drohendem Leberkoma, kurzfristige Eiweiß-Karenz; nach klinischer Besserung langsame Steigerung mit z.B. pflanzlichem EW oder Milcheiweiß.
→ II: Elektrolyt-/Säure-Base-Haushalt:
→ 1) Ausgleich einer Hypokaliämie oder Hyperkaliämie (Resonium/Hämodialyse).
→ 2) Ausgleich einer Hyponatriämie durch Wasserrestriktion (eine hypertone NaCl-Lösung führt zu einer Verdünnungshyponatriämie).
→ 3) Ausgleich einer Azidose bzw. Alkalose.
→ III: Bei Alkoholabhängigkeit intravenöse Thiamingabe.
→ IV: Bei GIT-Blutungen: Endoskopische Blutstillung durch Ligatur oder Sklerosierung, Gabe von FFP, Prothrombinkomplexkonzentrat, evtl. Erythrozyten- und/oder Thrombozytenkonzentrat.

→ V: Medikamentöse Therapie:
→ 1) Antibiotikagabe von Neomycin oder Rifampicin zu Reduktion der Ammoniak-bildenden Darmflora und/oder
→ 2) Laktulose: (3x 10-40ml/d). Es hemmt die bakterielle Urease und senkt somit die Ammoniakbildung; zum anderen senkt es den pH-Wert im Darm und vermindert die Diffusion von Ammoniak ins Blut (Lactulose ist ein nicht-resorbierbares Disaccharid, bestehend aus Galaktose und Fruktose. Es wird durch die Darmbakterien gespalten, wodurch Milchsäure entsteht, welche den pH-Wert des Darms senkt).
→ 3) Des Weiteren senkt die Gabe von L-Ornithin-Aspartat den Ammoniakspiegel im Körper und wirkt sich somit positiv auf die hepatische Enzephalopathie aus.
→ 4) Blutungsprophylaxe durch Gabe von Protonenpumpeninhibitoren (PPI wie Omeprazol, Pantoprazol).
→ 5) Die Gabe von Tranquilizern, Sedativa oder anderer hepatotoxischen Medikamente ist kontraindiziert.
→ 6) Bei Infektionen gezielte Antibiotikagabe durch Keimbestimmung (Blutkulturen).
→ 7) Das Hirnödem: Ist mit 70% die häufigste Komplikation der hepatischen Enzephalopathie. Hier erfolgt einer Oberkörperhochlagerung 45° sowie die Gabe einer 20%igen Mannitollösung intravenös (Erwachsene).
→ 8) Ultima ratio ist die Lebertransplantation und die extrakorporale Detoxikation mittels Promotheus oder MARS (= molecular-absorbent-recirculating-system). Letztere stellen Therapieoptionen zur Überbrückung der Wartezeit bis zur Lebertransplantation dar.