→ Definition: Beim Long-QT-Syndrom handelt es sich um eine kardiale Kanalopathie mit Verlängerung der QT-Zeit im EKG. Es geht mit einem erhöhten Risiko für Synkopen und plötzlichem Herztod durch Kammerflattern/-flimmern einher.
→ Ätiologie:
→ I: Angeboren: Es sind > als 10 Mutationen bekannt, jedoch > 85% betreffen das:
→ 1) LQT-1: Diese Form macht ca 40% der Fälle aus; hierbei sind die langsam-repolarisierenden Kalium-Kanäle (spannungsabhängig) betroffen durch einen Defekt der Alpha-Untereinheit des Ionenkanals, infolge einer Mutation des KCNQ1-Gens. Auslöser für die Rhythmusstörung sind u.a. körperliche Belastungen wie Schwimmen, Laufen, aber auch das Springen ins kalte Wasser.
→ 2) LQT-2: Betrifft die schnell-repolarisierenden Kaliumkanäle, infolge einer Mutation des HERG-Gens und konsekutivem Defekt der Alpha-Untereinheit dieses Ionenkanals.
→ 3) LQT-3: Hier besteht eine Mutation im SCN-5A-Gens mit einem Defekt in den spannungsabhängigen Natrium-Kanälen und einer Störung der Depolarisation (es kommt zur Überfunktion der Natriumkanäle; tritt in 6% der Fälle auf).

→ Klinisch-relevant: Das LQT-1-Syndrom lässt sich nochmals in 2 Subtypen unterteilen:
→ A) Jervell-Lange-Nielsen-Syndrom: Wird autosomal-rezessiv vererbt mit Verlängerung der QT-Zeit und angeborener Innenohrschwerhörigkeit.
→ B) Romano-Ward-Syndrom: Ist die häufigere kongenitale Form und wird autosomal-dominant vererbt; die Innenohrschwerhörigkeit fehlt.
→ II: Erworben: Hervorgerufen infolge einer medikamentösen Blockade des Kaliumstromes, durch
→ 1) Antiarrhythmika der Klasse I (Chinidin, Ajmalin, Lidocain oder Porpafenon) oder der Klasse III (Sotalol oder Amiodaron).
→ 2) Antidepressiva und Antipsychotika,
→ 3) Antimykotika: Ketoconazol, Fluconazol,
→ 4) Antihistaminika, etc.

→ Pathogenese: Diese Ionenkanalmutationen führen zu einer Veränderung entweder der Depolarisation (Natriumkanäle) oder der Repolarisation (Kaliumkanäle) und somit zu einer Verlängerung des Herz-Aktionspotenzials.
→ Klinisch-relevant: Bei Applikation von Medikamenten, die die QT-Zeit verlängern, immer:
→ A) EKG, und
→ B) Die Serumkaliumkonzentration überprüfen.
→ Klinik:
→ I: Charakteristische Beschwerden sind Palpitationen, Schwindel und Synkopen; Häufig weisen Kinder vor dem 12. Lebensjahr ein rhythmogenes Ereignis wie Torsades-de pointes -Tachykardie oder einen überlebten plötzlichen Herztod auf.
→ II: Die Symptome werden meist durch physischen oder psychischen Stress sowie durch Kälte ausgelöst.
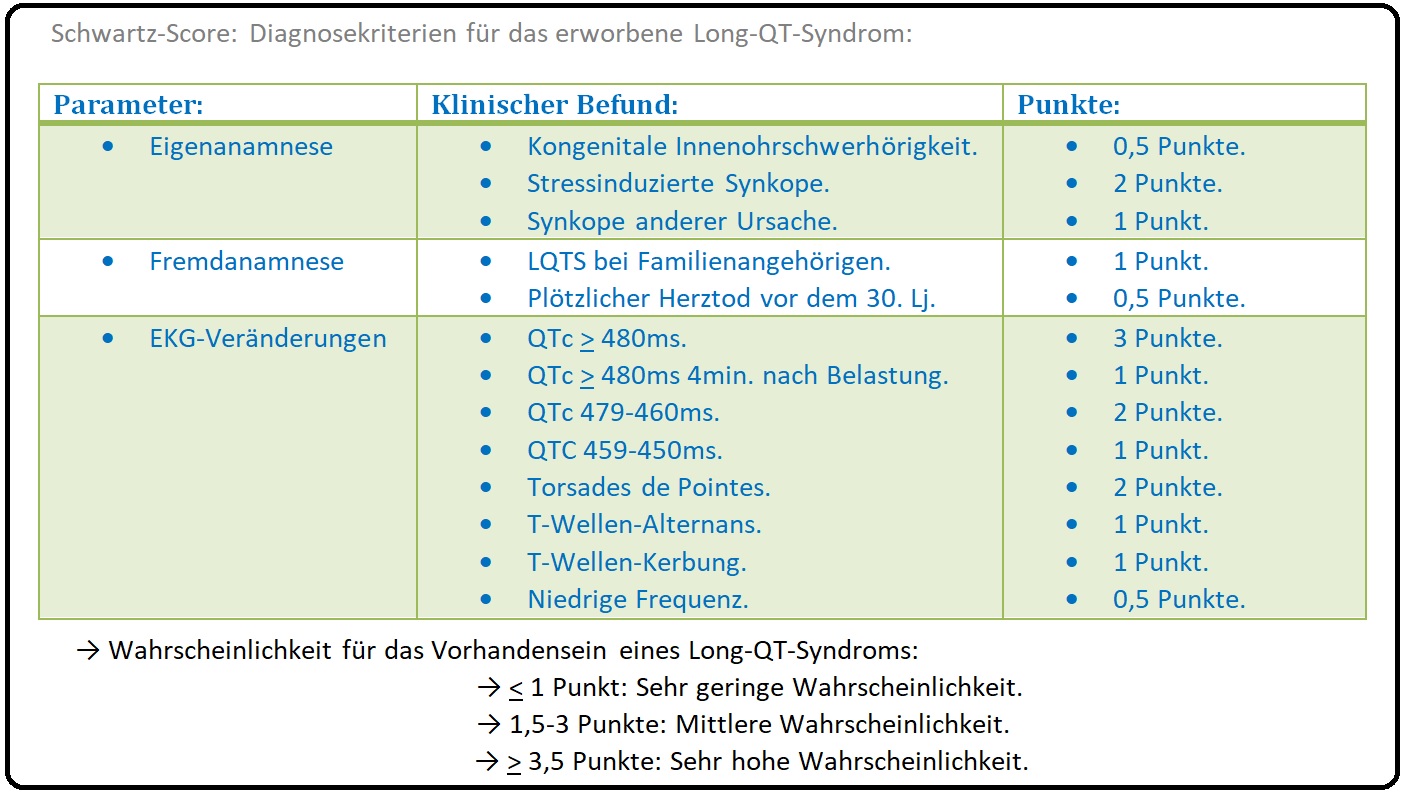
→ Diagnose:
→ I: Anamnese: Familienanamnese mit plötzlichen Todesfällen.
→ II: Ruhe-EKG: Bestimmung des korrigierten QTc-Intervalls (QT-Intervall in ms/ die Wurzel aus RR-Abstandes = QTc). Von einem verlängertem Aktionspotenzial spricht man:
→ 1) Bei Männern: QTc-Intervall > 440ms.
→ 2) Bei Frauen: QTc-Intervall > 470ms.
Mögliche Entwicklung einer Torsades-de-Pointes-Tachykardie mit konsekutivem elektrokardiographischem Nachweis (EKG-Befund: Torsades-de-pointes Tachykardie).
→ Therapie:
→ I: Bei der angeborenen Form sollten Triggersituationen wie Kälte, Sport, aber auch auditive Stimuli wie Wecker etc. gemieden werden.
→ II: Medikamente, die zu einer Hypokaliämie bzw. einer Verlängerung der QT-Zeit führen, sind kontraindiziert.
→ III: Prophylaktische Implantation eines ICD, sowie die Gabe eines ß-Blockers.
→ IV: Bei der erworbenen Form ist ein sofortiges Absetzten des auslösenden Medikamentes obligat, besteht eine Hypokaliämie wird Kalium substituiert.