→ Definition: Bei der idiopathischen thrombozytopenischen Purpura handelt es sich um eine hämorrhagische Diathese, die durch einen beschleunigten sowie durch Autoantikörper induzierten Abbau der Thrombozyten charakterisiert ist. Es existiert hierbei eine Stadieneinteilung zwischen:
→ I: Neu diagnostiziert: Bis zu 3 Monate nach Diagnosestellung; häufig zeigt sich eine Spontanremission.
→ II: Pesistierend: 3-12 Monate nach Diagnosestellung.
→ III: Chronisch: Mehr als 12 Monate nach Doagnosestellung.
→ Epidemiologie:
→ I: Der Morbus Werlhof stellt die häufigste immunologisch bedingte symptomatische Thrombozytopenie dar (ist insgesamt jedoch eine seltene Erkrankung).
→ II: Die chronische ITP tritt überwiegend im Erwachsenenalter auf, wobei Frauen im gebärfähigen Alter häufiger als Männer betroffen sind (F : M = 2,5 : 1).
→ III: Die akute Verlaufsform wiederum hat die höchste Inzidenz im Kindesalter (2-5 Jahre) und wird in über 60% der Fälle durch eine virale Infektion hervorgerufen.
→ Ätiopathogenese: Bei der idiopathischen thrombozytopenischen Purpura unterscheidet man zwischen 2 Verlaufsformen, denen beiden eine verminderte Plättchenzahl aufgrund eines vermehrten Thrombozytenabbaus und einer verminderten -überlebenszeit gemeinsam ist. Häufig ist die Ursache nicht zu eruieren:
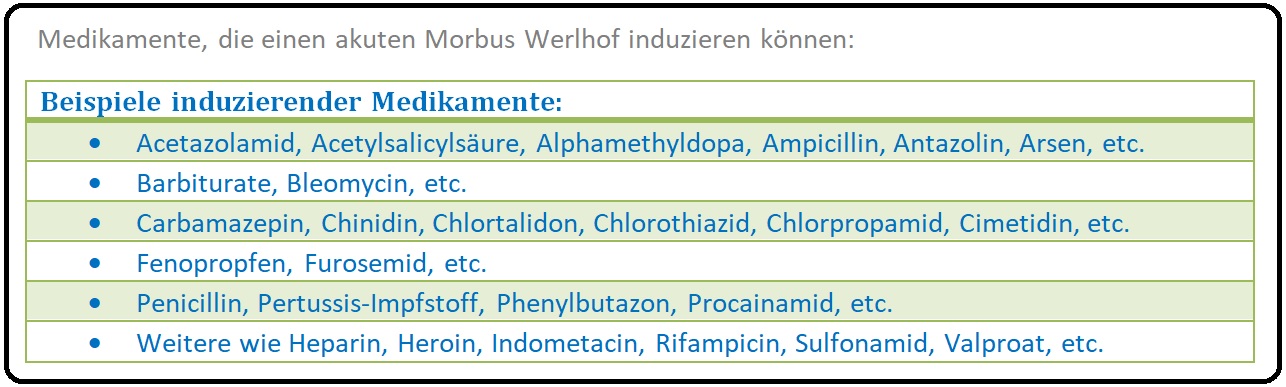
→ I: Akuter Morbus Werlhof: Tritt vorwiegend bei Kindern 5-10 Tage nach Exposition mit Viren (z.B. Mumps, Masern, Röteln, Virusinfektionen der oberen Atemwege, etc.) oder Medikamenten (z.B. Carbamazepin, Valproat, Furosemid, Heparin, Impfungen, etc.), Autoimmunerkrankungen, Antiphospholipid-Syndrom, etc.
→ 1) Charakteristischerweise binden Toxine oder Medikamente an ein Plasmaprotein, das zur Ausbildung von Antikörpern führt. Der neu gebildeten Antikörpern binden an die C3-Rezeptoren der Thrombozyten mit konsekutiv massivem Abfall der Plättchenzahl (Werte < 20000/µl).
→ 2) Auch im Knochenmark erfolgt die Bildung von Antikörpern, die zu einer Schädigung mit inadäquater Plättchenbildung (bei Megakaryozytose) führt.
→ II: Chronischer Morbus Werlhof: Von einer chronisch idiopathischen thrombozytopenischen Purpura spricht man bei einem Zeitintervall von mehr als 6 Monaten und es betrifft überwiegend Menschen im Erwachsenenalter. Diese Verlaufsform kann sich entweder aus der akuten Form entwickeln oder tritt spontan ohne Vor- oder Begleiterkrankungen auf.
→ III: Weitere Risikofaktoren: Sind u.a. Folgeerkrankung der chronischen B-Gastritis, etc.
→ Klinisch-relevant: Es wird angenommen, dass kreuzreaktive Antigene der viralen Erreger möglicherweise im Sinne einer molekularen Mimikry die Bildung von Antikörpern induzieren.
→ Klinik: Die klinische Symptome mit Petechien und Blutungen manifestiert sich zumeist ab einer Thrombozytenzahl < 20000/µl.
→ I: Akute ITP: Klinisch imponieren plötzlich auftretende Petechien insbesondere im Bereich der unteren Extremität, seltener an Stamm und oberer Extremität, Pupura, Nasenblutungen und Schleimhautblutungen, verstärktes Bluten nach Bagatelltrauma, Hämaturie bis hin zu schweren gastrointestinalen - und intrazerebralen Blutungen (letztere können terminal letal enden). Hinzu kommen nicht selten Erschöpfungssymptome.
→ II: Chronische ITP:
→ 1) Sie beginnt zumeist schleichend mit subkutanen Hämatomen, Nasen- und Schleimhautblutungen, Menorrhagien, etc..
→ 2) Im weiteren Krankheitsverlauf kommt es zu Petechien und Eckymosen bis hin zu schweren Blutungen.
→ III: Komplikationen: Wichtige z.T. schwerwiegende Komplikationen sind beim Morbus Werlhof u.a.:
→ 1) Thrombohämorrhagisches Syndrom mit Erythem, Zyanose und Grangrän.
→ 2) Evans-Syndrom: Hierbei handelt es sich um eine komplizierend hinzukommende autoimmunhämolytische Anämie.
→ Klinisch-relevant: Die chronisch idiopathische thrombzytopenische Purpura ist insbesondere mit lymphoproliferativen Erkrankungen wie die CLL und den Morbus Hodgkin, aber auch mit weiteren Erkrankungen wie HIV, Tuberkulose sowie Autoimmunerkrankungen z.B. systemischer Lupus erythematodes, Hashimoto-Thyreoiditis, etc. vergesellschaftet.
→ Diagnose:
→ I: Anamnese/klinische Untersuchung: Umfangreiche Anamnese von Medikamenten, Impfungen und möglichen vorangegangenen Infektionen.
→ II: Blutbild:
→ 1) Die Thrombozytenzahl kann zwischen 5000-100000/µl schwanken; insbesondere bei einer isolierten Thrombozytopenie sollte immer auch an eine idiopathische thrombozytopenische Purpura gedacht werden.
→ 2) Serologie: Möglicher Nachweis von Autoantikörpern (vom IgG-Typ) gegen die Thrombozyten-Glykoprotein-Komplexe GPIIb/GP IIIa, GPIb/IX/V bzw. GPIa/IIa sind in > 80% der Fälle nachweisbar aber nicht beweisend, da der Morbus Werlhof eine Ausschlussdiagnose darstellt.
→ III: Knochenmarkpunktion: Dient der differenzialdiagnostischen Abgrenzung und zeigt v.a. eine gesteigerte Megakaryozytopoese (bis auf das 5-fache der Norm).
→ Differenzialdiagnose: Vom Morbus Werlhof müssen insbesondere nachfolgende Erkrankungen abgegrenzt werden:
→ I: Alloimmunthrombozytopenie neonatal oder im Zusammenhang mit einer Transfusion von alloantikörperhaltigem FFP, aber auch thrombotische-thrombozytopenische Purpura.
→ II: Arzneimittel-induzierte Thrombozytopenie (z.B. Heparininduzierte Thrombozytopenie, etc.)
→ III: Myelodysplasien.
→ IV: Weitere Differenzialdiagnosen: Sind u.a.
→ 1) Hypersplenismus,
→ 2) Malaria und Leptospirose, etc.
→ Therapie: Bei einer Thrombozytenzahl > 50000/µl ist eine Therapie im Allgemeinen nicht nötig; erst bei Absinken der Thrombozyten < 20000/µl wird auch bei geringer Blutungsneigung eine Behandlung empfohlen.
→ I: Medikamentöse Therapie:
→ 1) Mittel der Wahl beim Morbus ist die Applikation von Prednisolon in einer Dosis von 1mg/kgKG/d über ein Zeitintervall von 1-2 Wochen. Zumeist kommt es zu einem Anstieg der Plättchen > 50000/µl, evtl. sogar zu einer Normalisierung. Im weiteren Therapieverlauf muss eine Erhaltungsdosis gefunden werden, bei der die Thrombozytenzahl > 20000-30000 persistiert. Liegt die Erhaltungsdosis < 10mg/d kann sollte die Behandlung über einen Zeitraum von 3-6 Monaten fortgeführt werden. Stellt sich hierbei aber keine Remission ein, besteht eine chronisch idiopathische thrombozytopenische Purpura und es sollte eine Splenektomie erwogen werden.
→ 2) Bei schweren z.T. lebensbedrohlichen Blutungen ist eine intensive Therapie, die zu einem raschen Thrombozytenzahl-Anstieg führt, obligat. Hier bietet sich eine Kombinationsbehandlung aus hochdosiertem Prednisolon (1g/d Methylprednisolon über 3 Tagen) und Immunglobulinen (400-1000mg/kgKG über 5 Tage) an. Zusätzlich kann bei dieser Therapie die Gabe von Thrombozytenkonzentraten nützlich sein. Die Transfusion von Thrombozyten sollte sehr zurückhaltend gestellt werden, da hierüber die Antikörperproduktion weiter stimuliert wird.
→ II: Operative Therapie: Bei der Splenektomie kommt es zur Beseitigung des Hauptabbauortes sowie zu einer Reduktion der antikörperbildenden Zellen. Zur Verringerung einer Postsplenektomiesepsis ist die präoperative Impfung der Betroffenen gegen Pneumokokken und Haemophilus obligat.
→ Prognose: Die Wahrscheinlichkeit einer Spontanremission liegt bei Patienten akutem Krankheitsbeginn bei bis zu 30-40%; demgegenüber ist sie bei Patienten mit schleichendem Krankheitsbeginn zumeist nur sehr gering. In 10% der Fälle kann die akute - in eine chronische Form übergehen.