→ Definition: Beim Rendu-Osler-Weber-Syndrom handelt es sich um eine autosomal-dominant vererbte Bindegewebserkrankung der Gefäße mit Manifestation von mukokutanen und viszeralen Angiodysplasien aufgrund einer Störung des Gefäßwandkollagens. Folgen sind u.a. Dilatation (Teleangiektasien im Bereich von Lippen, Mundhöhle, Nase, etc.) sowie Malformation kleinster Gefäße der inneren Organe (Lunge, Leber, GIT, ZNS) mit konsekutiver Entwicklung arteriovenöser Shunts.
→ Epidemiologie:
→ I: Die hereditäre hämorrhagische Teleangiektasie stellt mit einer Prävalenz von 1:2000-1:40000 eine eher seltene angeborene Vasopathie dar.
→ II: Geschlechterspezifische Unterschiede sind bis heute nicht bekannt.
→ III: Teleangiektasien und Epitaxis bestehen zumeist schon vor dem 20. Lebensjahr, eine viszerale Beteiligung findet man häufig erst 2-3 Dekaden später.
→ Ätiologie:
→ I: Es manifestiert sich hierbei ein Defekt des Endoglin-Gens auf Chromosom 9q3 bzw. der Aktivin-Rezeptor-ähnlichen-Kinase auf Chromosom 12q13.
→ II: Homozygote Formen sind nicht lebensfähig, heterozygote sind klassische Merkmalsträger; die Erkrankung ist an die Blutgruppe 0 gekoppelt.
→ Klassifikation: Je nach Mutation werden 2 Subtypen unterschieden:
→ I: HHT-1: Mutation im Endoglin-Gen. Klinisch manifestiert sich insbesondere ein Befall von Lunge und Gehirn.
→ II: HHT-2: Mit Mutation der Aktivin-Rezeptor-ähnlichen-Kinase (ALK1). Klinisches Korrelat ist der Befall von Leber und GIT.
→ Pahogenese: Gerade bei der HHT-1 ist bekannt, dass das Endoglin ein Membranglykoprotein an den Endothelzellen kodiert, welches an TGF-Beta (= Transforming-Growth-Faktor) bindet. Infolge dieses gestörten Gefäßbindegewebes entwickeln sich fokale Erweiterungen, insbesondere der postkapillären Venolen, die sich in die Kapillaren und Arteriolen fortsetzen. Im weiteren Krankheitsverlauf bilden sich aufgrund der Rarefizierung des Kapillarbettes, der Durchmesserzunahme und Elongation der Venolen Kurzschlussverbindungen mit den präkapillären Arteriolen aus. Charakteristikum der sich entwickelnden, mukokutanen Teleangiektasien ist eine ausgeprägte Vulnerabilität mit konsekutiver Blutungsneigung. Histologisch zeigt sich ein perivaskuläres Infiltrat mit mononukleären Zellen.
→ Klinik: Da die hereditäre hämorrhagische Teleangiektasie zahlreiche Organe betreffen kann, ist die klinische Symptomatik häufig sehr vielgestaltig:
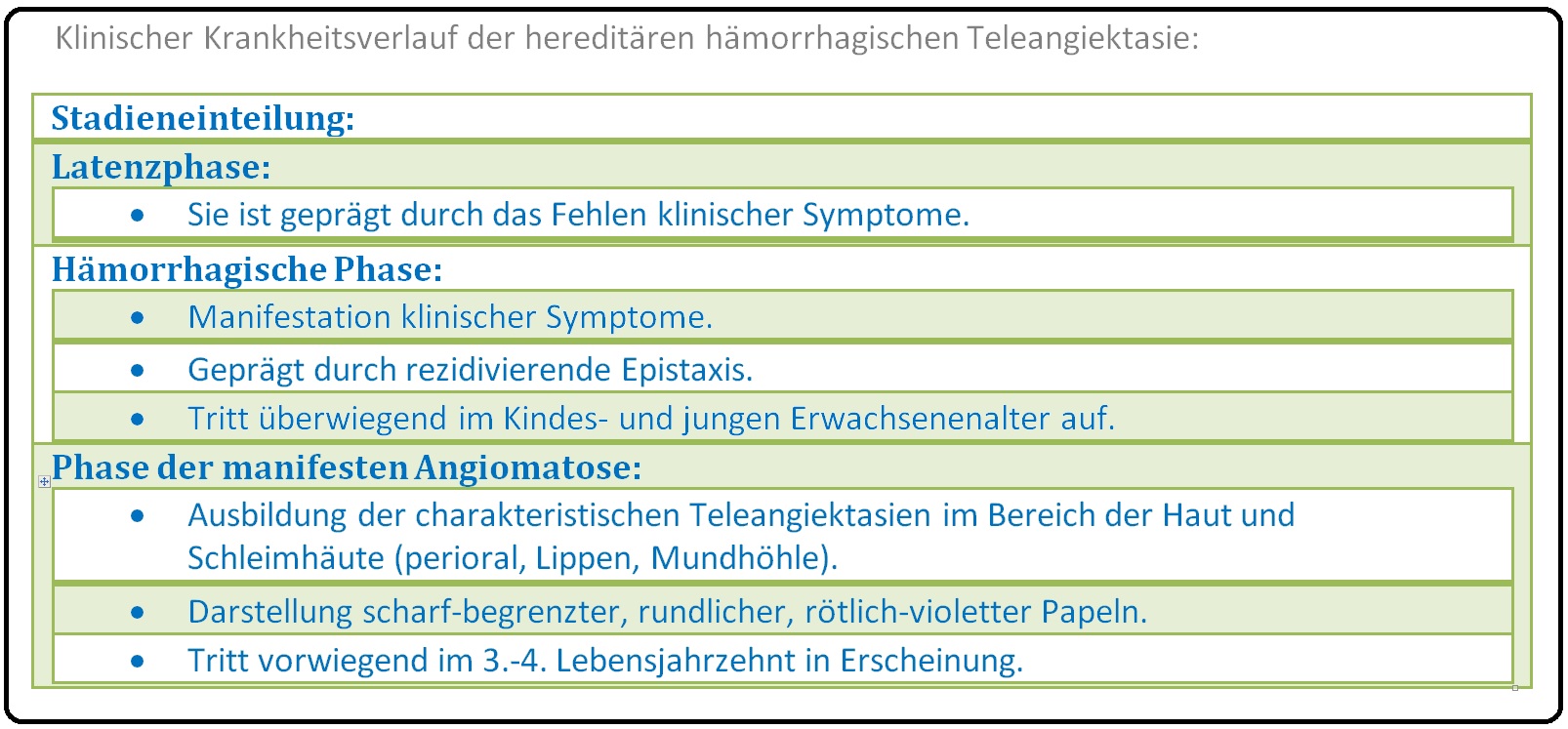
→ I: Der Morbus Osler weist einen stadienhaften Krankheitsverlauf auf. Erste klinische Symptome sind Epistaxis und Zahnfleischbluten, die bereits in der Kindheit und jungem Erwachsenenalter auftreten.
→ II: Im weiteren Krankheitsverlauf sind stecknadelkopf- bis reiskornkgroße rote Flecken an Haut und Schleimhäuten, insbesondere im Bereich des Gesichts, der Lippen, perioral, der Zunge, nasopharyngeal, an den Händen, aber auch im gesamten GI- und Urogenialtrackt nachweisbar. Diesbezüglich manifestieren sich u.a. Hämaturie, rezidivierende gastrointestinale Blutungen, chronische Eisenmangelanämie etc.
→ III: Weitere Symptome: Sind u.a.:
→ 1) Lungenbeteiligung: Manifestation von pulmonal arteriovenösen Malformationen und Shuntbildungen, die Thromben und Bakterien am Kapillarfilter der Lunge vorbeiführen und somit zu zerebralen und viszeralen Embolien sowie Abszedierungen (z.B. apoplektischer Insult, Hirnabszess etc.) führen. Des Weiteren können sich arteriovenöse Lungenfisteln entwickeln, die eine Symptomatik mit Dyspnoe, Hämoptysen, Zyanose, Trommelschlegelfinger bis hin zur pulmonalen Hypertonie und Herzinsuffizienz (= High-output-heart-failure) hervorruft.
→ 2) Leberbeteiligung: Findet man in bis zu 80% der Fälle mit asymptomatischen arteriovenösen Fisteln. Jedoch kann es durch Shuntbildungen zwischen der A. hepatica und Lebervenen bzw. Pfortader und Lebervenen zur portalen Hypertension mit Aszites, Ösophagusvarizen, Splenomegalie bis hin zur Leberzirrhose kommen.
→ 3) ZNS-Beteiligung: Infolge der zerebralen Teleangiektasien, arteriovenösen Malformationen (AVM), Aneurysmen und kavernösen Angiomen können sich Migräne- und Krampfanfälle, Synkopen bis hin zur Apoplexie sowie Subarachnoidal- bzw. Intrazerebralblutungen manifestieren.
→ 4) In einigen Fällen besteht eine Assoziation zur familiären juvenilen Polyposis.
→ Diagnose:
→ I: Anamnese/Klinische Untersuchung: Hinweise auf rezidivierendes Nasenbluten, gastrointestinale Blutungen oder positive Familienanamnese. Nachweis von Teleangiektasien an Haut und Schleimhäuten.
→ II: Bildgebende Verfahren: Bei unklaren Blutungen kann nach Endoskopie eine Angiographie indiziert sein, bei der sich gerade im Versorgungsgebiet der A. mesenterica superior (Mesenterium und Dünndarmmukosa) ein Netz von angiomatösen Fehlbildungen zeigt. Zum Nachweis von pulmonalen bzw. hepatischen Malformationen können zur genauen Diagonstik evtl. hrCT, MRT, MR-Angiographie etc. eingesetzt werden.
→ Klinisch-relevant:
→ A) Die klinische Diagnose wird anhand der Curacao-Kriterien gestellt:

→ B) Des Weiteren ist beim Morbus Osler eine charakteristische Diagnosetrias, bestehend aus:
→ 1) Mukokutanen Teleangiektasien,
→ 2) Erhöhter Blutungsneigung und
→ 3) Positiver Familienanamnese nachweisbar.
→ Differenzialdiagnose: Hiervon abzugrenzen sind u.a.:
→ I: CREST-Syndrom: Die klinische Symptomatik beinhaltet, Teleangiektasien, Raynaud-Syndrom, Calcinosis cutis, Sklerodaktylie und ösophageale Dysfunktion.
→ II: Angiodysplasien: Im Gegensatz zu den Angiodysplasien weist die hereditäre hämorrhagische Teleangiektasie pathologische Wandverdickungen der betroffenen Venolen sowie eine ausgeprägte longitudinale Muskulatur auf.
→ III: Morbus Fabry.
→ IV: Weitere Differenzialdiagnosen: Sind insbesondere:
→ 1) Leberzirrhose (als Differenzialdiagnose eines knotigen Leberumbaus).
→ 2) Caroli-Syndrom (hierbei sind die Gallenwege, nicht jedoch die intrahepatischen Äste der A. hepatica dilatiert).
→ 3) Fokal noduläre Hyperplasie (mit auffällig arterieller Vaskularisation).
→ Therapie: Aufgrund der extremen klinischen Variation besteht bei der HHT keine spezifische kausale Therapie, vielmehr gestaltet sie sich interdisziplinär mit besonderem Augenmerk auf die symptomatische Therapie bzw. Prävention.
→ I: Konservative Therapie: Bei bestehender Herzinsuffizienz aufgrund von arteriovenösen oder arterioportalen hepatischen Shunts können Beta-Blocker zur Behandlung appliziert werden. Zur Steuerung der medikamentösen Therapie bei symptomatischen Patienten ist die Bestimmung des Herzzeitvolumens z.B. mittels Echokardiographie vor Therapiebeginn obligat. Zudem sind eine Salzrestriktion sowie diuretische Therapie indiziert. Weitere konservative Maßnahmen umfassen:
→ 1) Nasenbluten: Zur Prophylaxe von Nasenbluten wird eine fetthaltige Nasensalbe empfohlen. Bei rezidivierender Epistaxis kann zur lokalen Blutstillung ein vasokonstriktives Nasenspray appliziert werden.
→ 2)Teleangiektasien: Mukokutane Teleangiektasien können bei Blutungskomplikationen (z.B. gastrointestinale Blutungen) mittels Laserkoagulation, septaler mukosaler Dermatoplastie oder selektiver Embolisierung von Gefäßen therapiert werden.
→ 3) Anämie: Bei Auftreten einer chronischen Blutungsanämie aufgrund von rezidivierenden gastrointestinalen, urogenitalen etc. Blutungen kann bei leichteren Formen Eisen substituiert werden, bei schwereren Formen ist ein Bluttransfusion indiziert.
→ II: Interventionelle Therapie: Arteriovenöse Malformationen werden zumeist mittels Embolisation oder stereotaktischer Radiotherapie, seltener durch mikrochirurgische Exzision therapiert. Gerade bei den zerebralen Malformationen werden kleinere Angiodysplasien (< 3cm) mittels stereotaktischer Radiotherapie, größere (> 3cm) mittels Embolisation behandelt. Auch bei den hepatischen und pulmonalen Teleangiektasien erfolgt zumeist eine Embolisation, wobei insbesondere die pulmonale Form eine frühzeitige Indikation infolge der häufig beobachteten Größenprogredienz (> 3cm) erfordert, während die asymptomatische, hepatische Form primär nicht therapiert wird. Hier sind regelmäßige Kontrollen der Leberfunktion obligat.