→ Pharmakokinetik: (Siehe auch Antidepressiva allgemein):
→ I: Antidepressiva werden meist oral verabreicht und haben aufgrund ihrer ausgeprägten Lipophilie und Plasmaproteinbindung ein hohes Verteilungsvolumina.
→ II: Die Bioverfügbarkeit ist jedoch durch den hohen First-pass-Effekt der Leber gering.
→ III: Eine parenterale Applikation ist gerade bei den trizyklischen/tetrazyklischen AD sowie bei Mirtazapin möglich.
→ Klinisch-relevant:
→ I: Der maximale Plasmaspiegel bei den AD wird nach 1-6 Stunden erreicht.
→ II: Die Eliminations-HWZ der ANtidepressiva liegt zwischen 10-40 Stunden.
→ III: Ausnahme:
→ 1) Präparate mit sehr kurzer Halbwertszeit (= HWZ) sind z.B.:
→ A) Venlafaxin mit einer sehr kurzen HWZ von 5 Stunden und
→ B) Trazodon mit 6 Stunden.
→ 2) Eine sehr langen HWZ von ca. 2-7 Tagen hat Fluoxetin.
→ IV: Der Steady-state (= Der Zeitpunkt bei dem sich die resorbierten und die eliminierten Medikamentenmengen die Waage halten = Fließgleichgewicht) der Antidepressiva wird nach 5-10 Tagen erreicht.
→ Metabolisierung/Elimination:
→ I: Die Metabolisierung der Antidepressiva erfolgt hepatisch u.a. durch Demythelierung, sodass z.T. aktive Metaboliten (Imipramin zu Desmipramin = Desmethylimipramin) entstehen.
→ II: Weitere Metabolisierungsschritte der Antidepressiva sind u.a. Oxidation und Konjugation von Glucuronsäuren in der Leber.
→ III: Die Ausscheidung erfolgt fast ausschließlich renal.
→ Biochemie:
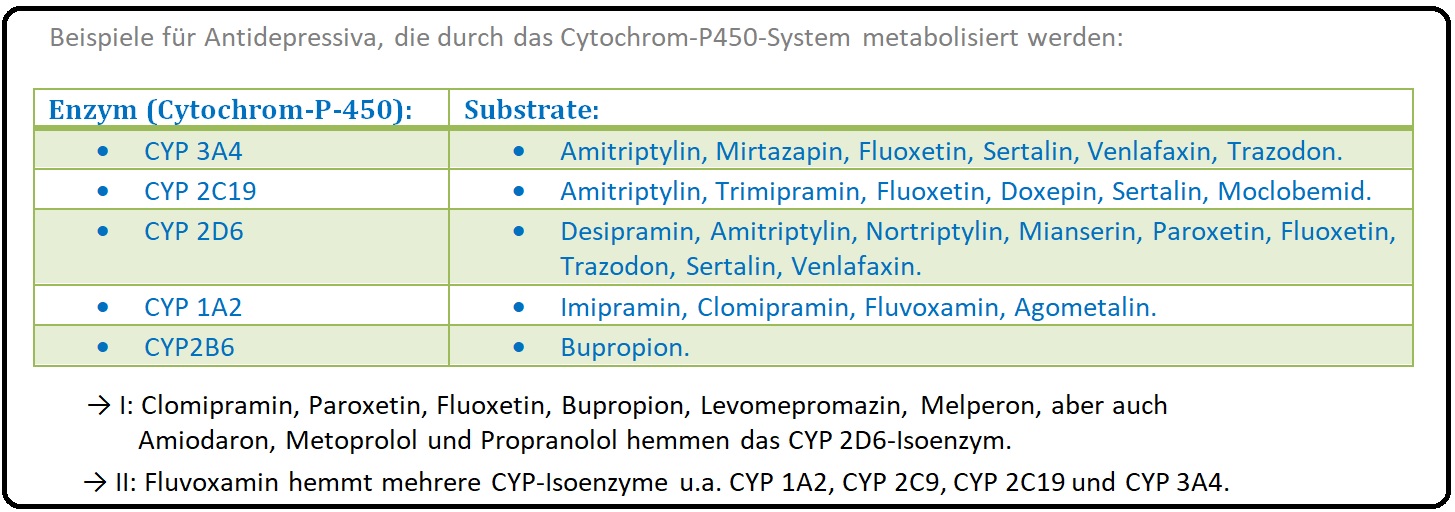
→ I: Cytochrom-P-450-System: Die Verstoffwechselung fast aller Psychopharmaka (Ausnahme: Lithium und Amisulprid) erfolgt über das Cytochrom-P-450-System. Die wichtigsten Enzyme für diese Psychopharmaka sind:
→ 1) CYP-1A2,
→ 2) CYP2C9,
→ 3) CYP2C19,
→ 4) CYP2D6,
→ 5) CYP3A4, etc.
→ II: Bei 10% der Allgemeinbevölkerung findet man gerade bei den Enzymen CYP2A6, CYP2C9, CYP2C19 sowie CYP2D6 einen Polymorphismus, der dazu führt, dass die Enzymaktivität sehr stark oder sehr schwach bis fehlend ist. Hierdurch werden Medikamente besonders schnell oder langsam metabolisiert. Man unterscheidet:
→ 1) Ultra-rapid Metabilizer: Diese bauen das Pharmakon besonders schnell ab, sodass kein ausreichender Plasmaspiegel erreicht wird.
→ 2) Poor-Metabolizer: Das Pharmakon wird nur sehr langsam abgebaut, sodass nur geringe Mengen schon zu schweren Nebenwirkungen bzw. Intoxikationen führen können.
→ 3) Extensive Metabolizer: Hierbei wird das Pharmakon normal abgebaut, es besteht kein Polymorphismus.
→ III: Einige Beispiele: Welche Cytochom-P-450-Enzyme, welche Antidepressiva metabolisieren.
→ 1) CYP1A2: Amitriptylin, Imipramin, Mirtazapin.
→ 2) CYP2C9: Amitryptilin, Fluoxetin, Sertralin.
→ 3) CYP2C19: Amytriptilin, Trimipramin, Doxepin, Imipramin, Citalopram.
→ 4) CYP2D6: Amitriptylin, Desipramin, Nortyptilin, Fluoxetin.
→ IV: Polymorphismus: Diese kann mittels PCR (= Polymerase-Kettenreaktion) nachgewiesen werden. Gerade bei dem CYP2D6 liegen bei der Allgemeinbevölkerung (10%) mehrere Gene vor, sodass es zu einer gesteigerten Duplikation mit einem vermehrten Abbau des Pharmakons kommt (Ultra-rapid-Metabolizer).
→ V: Weitere polymorphe Cytochrom-P-450-Isoenzyme:

→ Klinisch-relevant:
→ A) Gerade die älteren SSRI wie Fluoxetin, Paroxetin, Fluvoxamin können die Cytochom-P-450-Enzyme hemmen und somit zu einer Erhöhung des Plasmaspiegel mit Gefahr der Intoxikationen führen.
→ B) Carbamazepin, Rifampicin, Johanniskraut und Rauchen können die P-450-Enzyme induzieren und somit einen schnelleren Abbau von Carbamazepin selbst (Selbstinduktion), aber auch von Amitriptylin, etc. verursachen.
→ Wechselwirkungen:
→ I: Gerade die Antidepressiva weisen mit anderen pharmazeutischen Substanzen ausgeprägte Wechselwirkungen auf, da durch das Cytochrom-P-450-Systems eine komplexe Medikamenteninteraktion besteht.
→ 1) CYP1A2: Clozapin:
→ A) Induktion durch Carbamazepin/Rauchen mit konsekutiver Erniedrigung der Clozapin-Konzentration.
→ B) Hemmung der CYP-1A2 durch Fluvoxamin mit Erhöhung der Clozapin Konzentration.
→ 2) CYP2C9: Phenytoin, Phenprocoumon:
→ A) Carbamazepin und Rifampicin führen durch Induktion zur Erniedrigung der Pharmakons,
→ B) Fluoxamin und Valproinsäure führen wiederum zur Erhöhung der Konzentration.
→ II: Trizyklische AD:
→ 1) Interaktion mit Alkohol: Verstärkung der zentral-dämpfenden Wirkung.
→ 2) Interaktion mit Anticholinergika und Antihistaminika; sie verstärken die cholinergen Nebenwirkungen wie Engwinkelglaukom, Darm- und Blasenatonie und Delir.
→ 3) Interaktion mit Neuroleptika; hierbei besteht eine gegenseitige Verstärkung mit konsekutiver Steigerung der Plasmaspiegel.
→ 4) Interaktion mit MAO-Hemmern; evtl. starke Blutdruckschwankungen, Fieber, Tremor, Erregungszustände, Übelkeit, Erbrechen.
→ 5) Interaktion mit Antiphypertensiva: Wie Clonidin führen zur verminderten blutdrucksenkenden Wirkung.
→ 6) Interaktion mit Sympathomimetika: Wie Adrenalin führen zur Blutdrucksteigerung.
→ II: SSRI:
→ 1) Antikoagulantien: Erhöhte Blutungsgefahr;
→ 2) Serotonergen Stoffen: Triptan-Migränemittel: Serotonin-Syndrom.
→ 3) Trizyklische AD: Erhöhung des TZA-Plasmaspiegels.
→ Klinisch-relevant:
→ I: Bei starken Rauchern kann die plötzliche Abstinenz (Zigarettenrauch, ein Cytochrom-P-450-Enzym-Induktor, verursacht einen rascheren Abbau des Pharmakons) zu einem Anstieg der Plasmakonzentration führen und konsekutiv Nebenwirkungen verstärken bzw. eine Intoxikation induzieren.
→ II: Plasmakonzentration: Die regelmäßige Kontrolle im Sinne des therapeutischen-Drug-Monitoring (= DTM) ist von großer Bedeutung und sollte erst nach Erreichen des Steady-state-Zeitpunktes indiziert sein. Dies ist ca. nach 4 Halbwertszeiten möglich.
→ III: Das therapeutische-Drug-Monitoring ist insbesondere bei Patientengruppen, z.B. Schwangeren, Kindern, älteren Menschen mit geringer/fehlender Compliance, sowie bei Patienten mit einer Variation des Cytochrom-P-450-Systems obligat.