→ Definition: Bei der Mukoviszidose handelt es sich um eine autosomal-rezessive Stoffwechselkrankheit, die auf einer Störung des transmembranösen Elektrolyttransportes der Epithelien von Schleimhäuten und Drüsengängen (exokrinen Drüsen) beruht. Sie manifestiert sich insbesondere an:
→ I: Lunge: Obstruktion des Bronchialsystems aufgrund eines eingedickten Bonchialsekrets mit erhöhtem Risiko für bronchopulmonale Infekte.
→ II: Pankreas: Mit Insuffizienz der exokrinen Pankreasfunktion.
→ III: GIT: Mit Malabsorption und Störungen der Motilität sowie
→ IV: Weiteren Störungen wie Cholestase (hepatobiliäres System) und Infertilität (Urogenitalsystem).
→ Epidemiologie: Die Mukoviszidose stellt die häufigste angeborene Stoffwechselerkrankung in Mittel- und Westeuropa dar. In Europa kommt ein Erkrankungsfall auf etwa 2500 Geburten.
→ Ätiopathogenese: Die Mukoviszidose wird autosomal-rezessiv vererbt mit Mutationen auf dem langen Arm des Chromosom 7. Genprodukt ist ein Protein, das den transmembranösen Elektrolyttransport reguliert (bildet einen c-AMP-abhängigen Chloridkanal) und wird als CFTR-Protein (= Cystic-Fibrosis-Transmembrane conductance-Regulator) bezeichnet.
→ I: Klassifikation: Aufgrund der vielen Mutationen lässt sich die Mukoviszidose hinsichtlich der Auswirkungen in 4 Gruppen unterteilen:
→ 1) Mutation, die die Synthese des Proteins vollständig verhindern (irreguläres Stopp-Codon).
→ 2) Mutation, die zu einer falschen „Prozessierung“ des Proteins führen. Folge ist eine fehlerhafte Faltung oder inkorrekte Glykosylierung des Proteins, das schließlich intrazellulär abgebaut wird.
→ 3) Mutation, die eine physiologische Regulationsfunktion verhindert z.B. durch Veränderung der Bindungsstelle für ATP.
→ 4) Mutation, die den transmembranösen Elektrolytfluss beeinflussen, jedoch nicht gänzlich verhindern z.B. durch Änderung der Aminosäurensequenz des Ionenkanals.
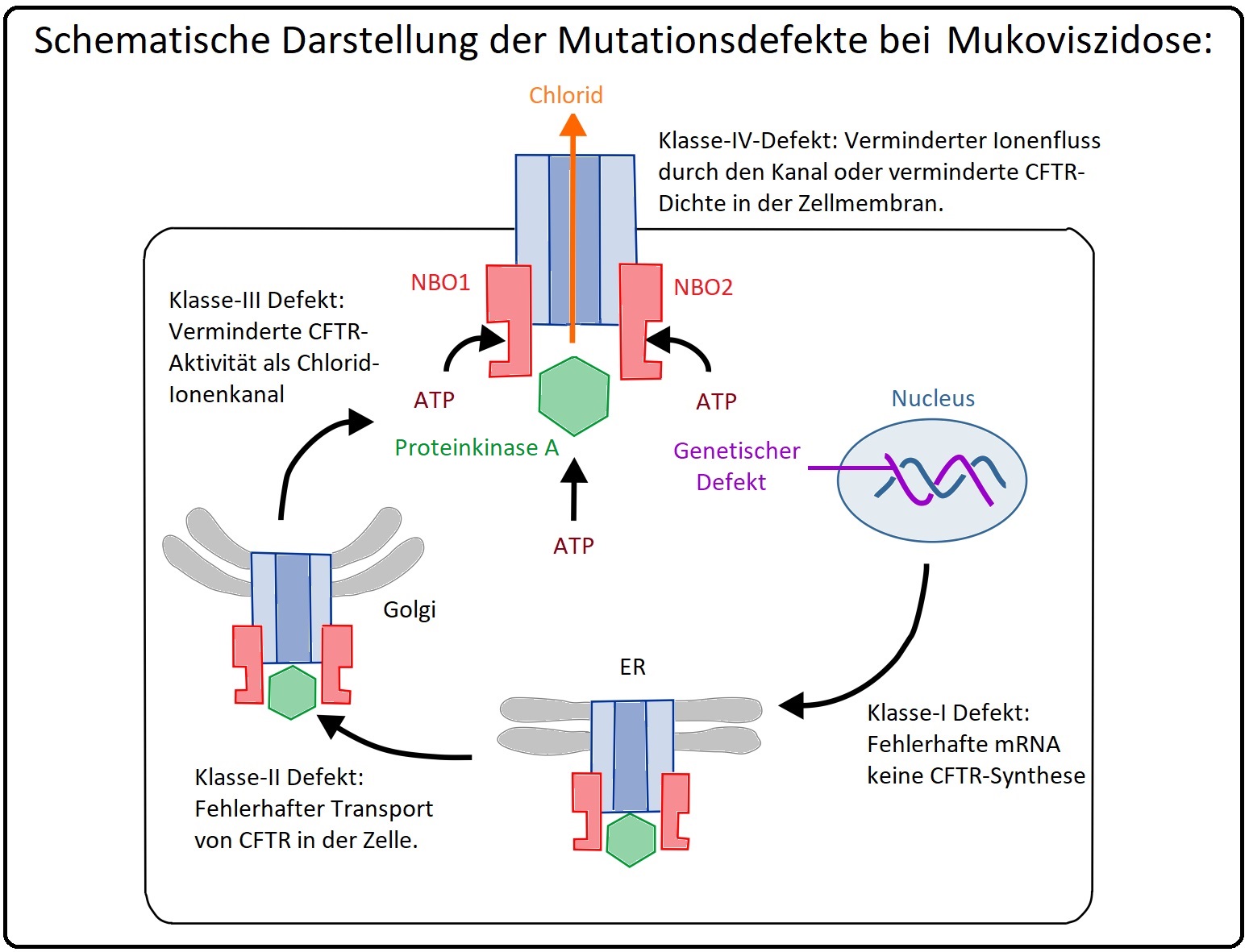
Die häufigste Mutation bei der Mukoviszidose ist jedoch eine Deletion von 3 Basen an Position 508 mit Ausfall des Phenylalanins.
→ II: Das CFTR-Protein hat die Funktion eines transmembranösen cAMP-regulierten Chloridkanals. Zudem ist es Regulator weiterer transmembranöser Ionenkanäle wie den basolateralen K+-Kanäle und den apikalen Na+-Kanäle in den Epithelien. Auch im Bereich der Exo- und Endozytose sowie der Bildung von sekundären Signalstoffen weist das CFTR-Protein eine regulatorische Funktion auf.
→ III: Pathogenese: Das CFTR-Protein ist in der apikalen Epithelmembran lokalisiert und stellt einen cAMP-regulierten Chloridkanal dar. Zudem variiert die Funktion des CFTR-Proteins in Abhängigkeit mit dem Standort des Epithels:
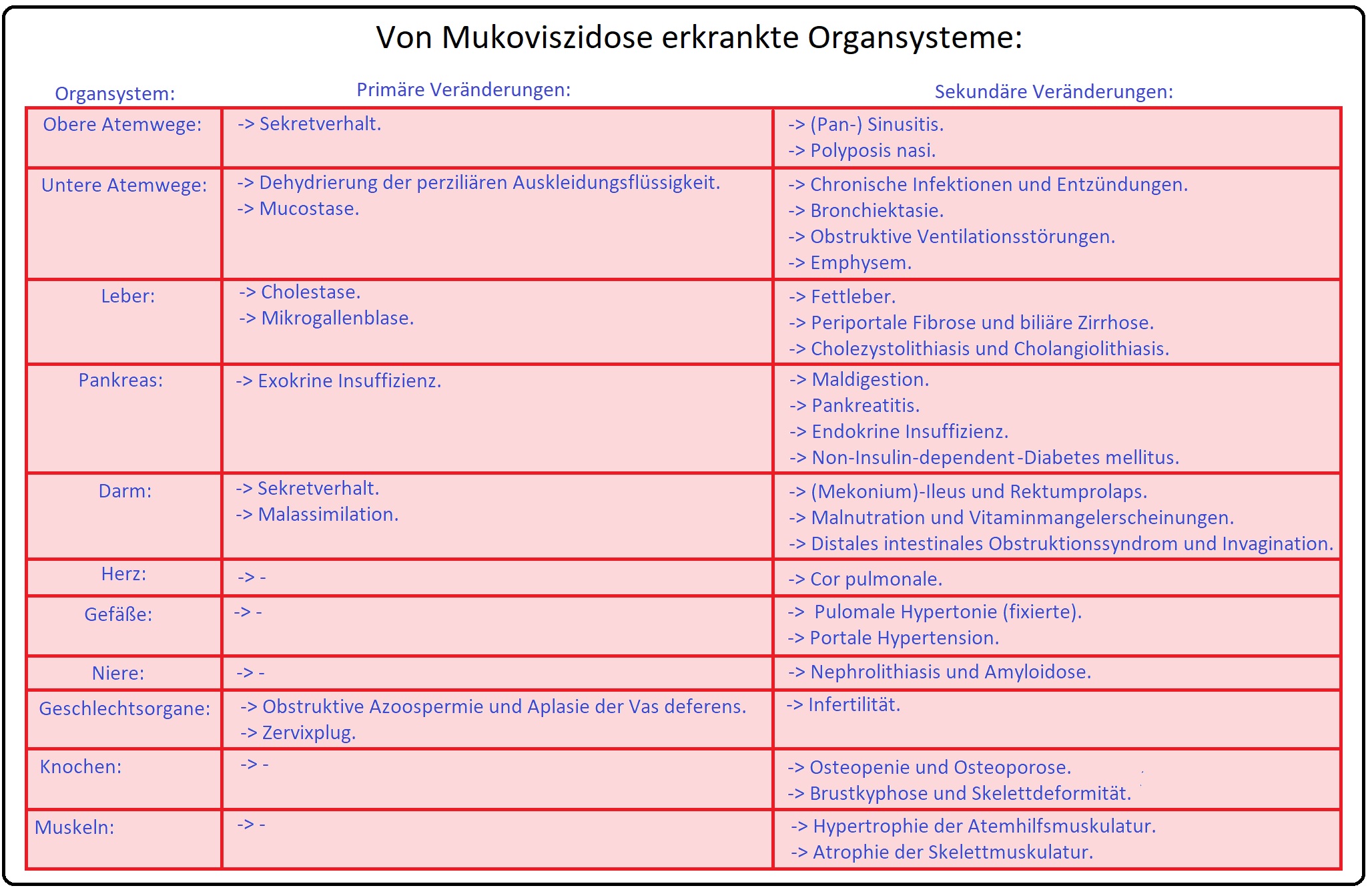
→ 1) Atemwege: Der CFTR-Defekt beschleunigt die Wasser und Na+-Resorption des Epithels und hemmt die Cl--Sekretion in das Bonchialsekret. Folge ist ein viköses, klebriges Sekret, das nicht mehr durch die Ziliarbewegung oder Husten aus den kleinen Bronchen entfernt werden kann. Charakteristischerweise kommt es zur chronischen Obstruktion durch Sekretstau und sekundären Infektionen mit konsekutiver Destruktion des Lungenparenchyms.
→ 2) Pankreas: Das Defekt der cAMP-abhängigen Chloridkanäle in den pankreatischen Ductuli hemmt die Chlorid und konsekutiv auch die Bikarbonat-Sekretion sowie die passive Na+ und Wassersekretion. Es resultiert eine Eindickung des Pankreassekrets mit Ductuli-Okklusion mit Destruktion des Pankreasparenchyms. Klinisches Korrelat ist die exkretorische Pankreasinsuffizienz.
→ 3) Intestinaltrakt: Im oberen Dünndarm ist durch die eingeschränkte Na+- und Wassersekretion die Ausschwemung von Muzinen und Makromolekülen aus den Krypten vermindert. Im distalen Dünndarm und im Kolon manifestiert sich eine abnorm gesteigerte Na+- und Wasserrückresorption und führt nicht selten schon bei Neugeborenen durch Eindickung des Kots zum Mekoniumileus.
→ 4) Genitalsystem: Bis zu 95% der Männer weisen eine Azoospermie infolge einer Obliteration der Vas deferens durch eingedicktes Sekret auf.
→ Klinik: Zumeist beginnt die Erkrankung unmittelbar nach der Geburt oder im Kleinkindesalter (nur bei 7% der Fälle wird die Diagnose nach dem 18. Lebensjahr gestellt).
→ I: Respirationstrakt:
→ 1) Leitsymptom ist der Husten, der bei bakterieller Infektion purulentem Auswurf (häufige Erreger sind insbesondere Haemophilus influenzae, Pseudonomas aeroginosa, Staphylococcus aureus, etc.) aufweist und sich im weiteren Krankheitsverlauf chronifiziert; es entwickelt sich eine zunehmende obstruktive Bronchitis mit konsekutiver Abnahme der Lungenfunktion. Klinisches Korrelat ist u.a. Ruhetachypnoe, Orthopnoe und generelle Zyanose, aber auch Belastungsintoleranz, Appetitverlust und Gewichtsabnahme.
→ Klinisch-relevant: Bei Absinken des pO2 im Blut unter 55mmHg manifestiert sich eine pulmonale Hypertonie mit konsekutivem Cor pulmonale.
→ 2) Der Endzustand ist die (partielle) pulmonale Insuffizienz mit Entwicklung von z.B. Trommelschlägelfinger und hypertrophen pulmonalen Osteopathie, aber auch Komplikationen wie Pneumothorax und/oder Hämoptysen. Weitere Symptome sind chronische Sinusitis und Polypenbildung; auffällig selten ist die Pleura betroffen.
→ II: Pankreas: Häufige klinische Symptome sind u.a.:
→ 1) Exokrine Pankreasinsuffizienz mit Fettmaldigestion und Mangel an fettlöslichen Vitaminen (E, D, K und A).
→ 2) Auch die endokrine Pankreasfunktion kann bei der Mukoviszidose eingeschränkt sein; Es kommt zur Destruktion der Alpha- und Beta-Zellen und somit zur verminderten Insulin- und Gukagonausschüttung. Klinische Korrelat ist die verminderte Glukosetoleranz und das Auftreten eines Diabetes mellitus.
→ 3) Rezidivierende (akute) Pankreatitiden.
→ III: Hepatobiliäre System: Initiales Symptom ist die Cholestase im weiteren Krankheitsverlauf manifestieren sich cholestatische Komplikationen mit vermehrten Gallengangssteinen (Kalziumbilirubinat) bis hin zur portalen Hypertension und Leberzirrhose.
→ IV: Ein distal intestinales Obstruktionssyndrom wird durch Verlegung des terminalen Ileums mit voluminösen Stuhl bedingt.
→ V: Schweißdrüsen: Die Dysfunktion ist ein wichtiges Symptom für die Diagnostik der Erkrankung. Sowohl die Schweißmenge als auch die Konzentration von Na+, K+ und Cl- sind deutlich erhöht. Mögliche Folgen sind u.a.:
→ 1) Salzverlust mit Hypotonie und Hypochlorämie und seltener
→ 2) Hyponatriämie mit Alkalose.
→ VI: Genitaltrakt:
→ 1) Männern: Verspätete Pubertät sowie häufig eine Infertilität. Sie beruht auf einer Störung des Elektrolyttransportes im Epithel der Samenblase und Samenleiter mit konsekutiver Veränderung des Spermas und einer Azoospermie trotz testikulärer Spermiogenese.
→ 2) Frauen: Bei den Frauen ist die Menarche verspätet und die Fertilität eingeschränkt. Es zeigen sich klinische Symptome wie Menstruationsstörungen, Oligomenorrhö und Schleimverschluss der Cervix uteric, etc.
→ Diagnostik:
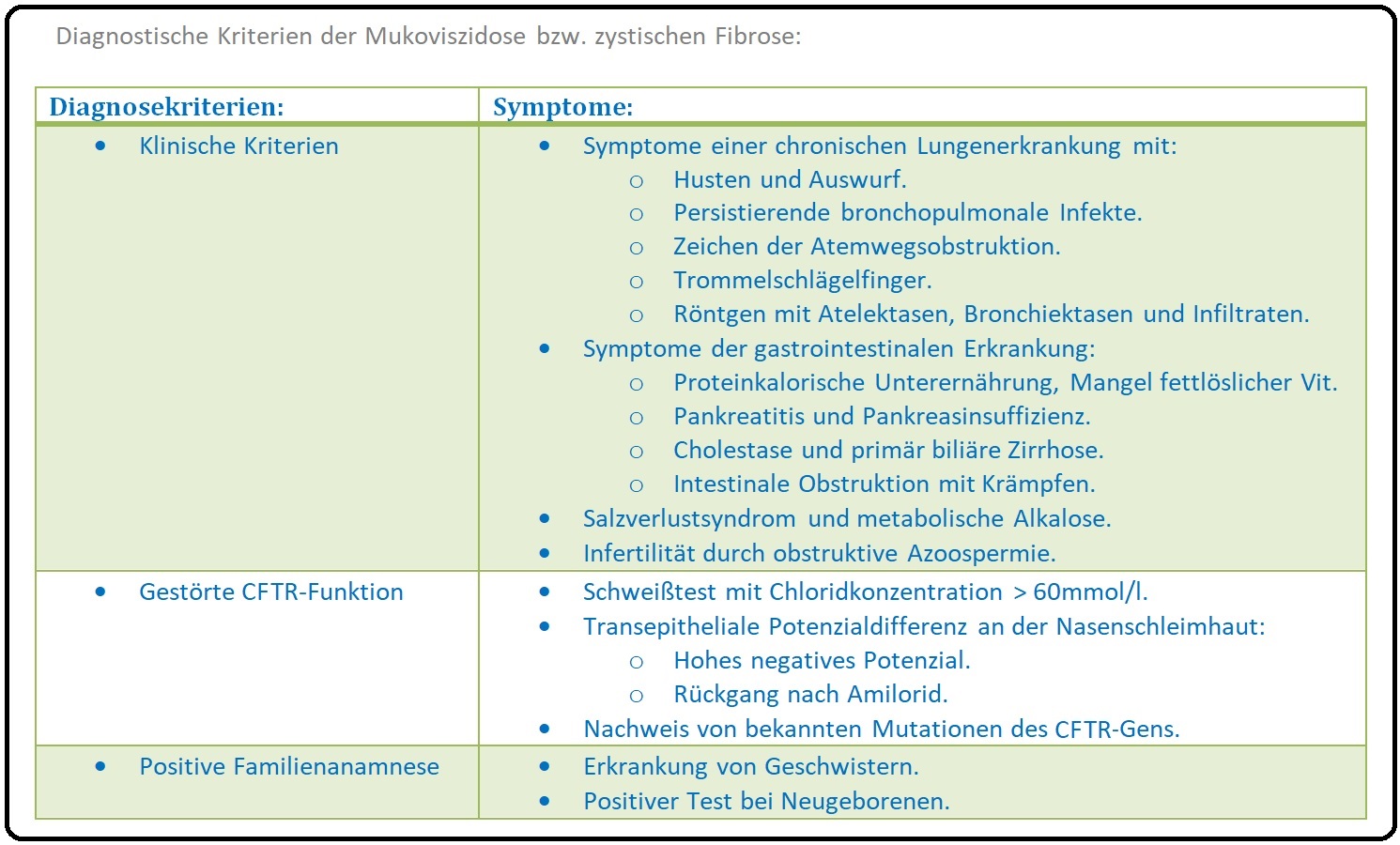
→ I: Anamnese/klinische Untersuchung: Bei Neugeborenen Unvermögen zu schreien, aufgeriebener Leib, ausbleibende Darmentleerung. Später ist dann die charakteristische Kombination aus respiratorischen Symptomen (rezidiverende bronchopulmonale Infekte wie Pneumonien, Fassthorax, Hypertrophie der Atemhilfmuskulatur) und gastrointestinalen - wegweisend.
→ II: Weitere klinische Untersuchungen:
→ 1) Lungenfunktionstest: Initial Zunahme des Residualvolumens im Vergleich zur Totalkapazität sowie Verminderung des exspiratorischen Volumens bei forcierter Ausatmung. Im weiteren Krankheitsverlauf Entwicklung einer partiellen pulmonalen Insuffizienz mit Absinken des pO2 - und schließlich des pCO2-Wertes. Insbesondere korrelieren die Vitalkapazität (VC) als auch die Einsekundenkapazität (FEV1) mit dem Stadium der Lungenerkrankung.
→ 2) Radiologischer Nachweis von evtl. bestehenden Atelektasen, Bronchiektasen und weiteren Infiltraten, etc.
→ III: Schweißtest: Nach Applikation und Stimulation mit Pilocarpin wird die Chloridkonzentration im Schweiß gemessen. Eine Chloridkonzentration > 60mmol/l bei zwei getrennten Test gilt als beweisend für die Mukoviszidose.
→ IV: Goldstandard bei der Diagnose der exokrinen Pankreasinsuffizienz ist die quantitative Bestimmung des Fettgehaltes im Sammelstuhl unter protokollierter Fettaufnahme bzw. die Bestimmung der Pankreaselastase im Stuhl.
→ IV: Genetische Analyse mittels DNA.
→ Differenzialdiagnose: Die Kombination aus Lungen- und Pankreasveränderungen sollte immer an eine Mukoviszidose denken lassen. Jedoch müssen zudem weitere Erkrankungen wie:
→ I: Chronische entzündliche Lungenerkrankungen wie COPD, Asthma bronchiale, etc. und
→ II: Pankreasinsuffizienz unterschiedlicher Genese ausgeschlossen werden.
→ Therapie: Eine kausale Therapie besteht bei der Mukoviszidose bis heute nicht, sodass sich die Behandlung insbesondere auf die Funktionsstörungen konzentriert.
→ I: Lunge:
→ 1) Das Abhusten des zähen Schleims kann durch physikalische Maßnahmen wie Vibrationen, Abklopfen und Training der Atemmuskulatur verbessert werden.
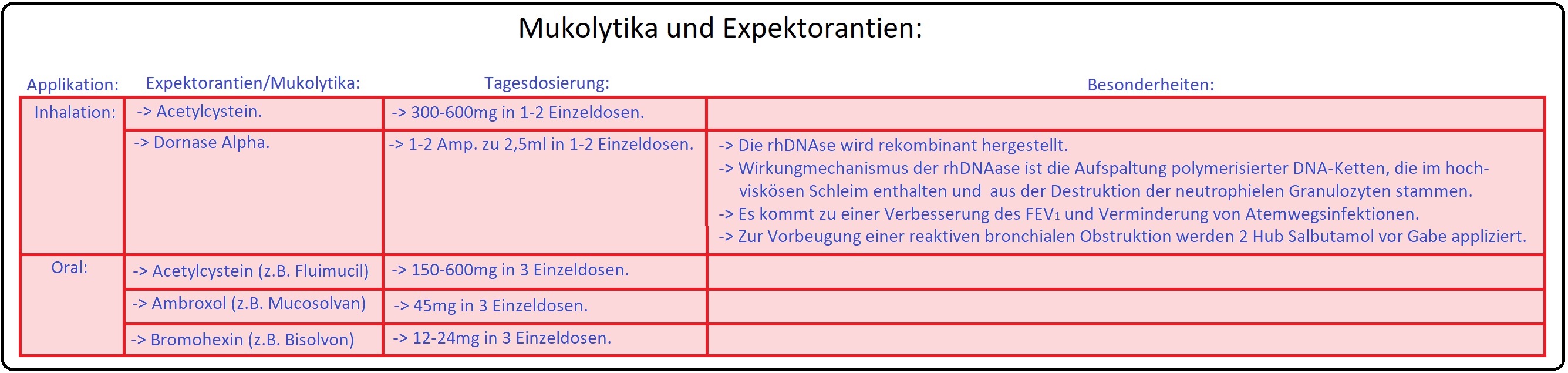
→ 2) Zur Sekretverflüssigung können auch Aerosole wie Kochsalzlösung (0,45%ig), Mykolytika wie N-Acetylcystein und Sekretolytika wie Ambroxol verabreicht werden.
→ 3) Bronchodilatatoren wie Beta2-Adrenergika sind nur kurzfristig bei bronchospastischen Zuständen indiziert. Bei langfristigem Gebrauch besteht die Gefahr der Hypertrophie submuköser Drüsen mit der Gefahr der verstärkten Schleimsekretion.
→ 4) Bei akuten entzündlichen Schüben sollte eine frühzeitige prolongierte Antibiotikatherapie nach Sputumkultur gezielt erfolgen. Bei leichter Infektionen mit Haemophilus influenzae ist z.B. ein Penicillin oder Cephalosporin per os indizert. Bei schwerer Infektionen mit z.B. Pseudonomas aeroginosa müssen Aminoglykoside einem weiteren Antibiotika (z.B. Cephalosporin) intravenös appliziert werden.
→ 5) Weitere Interventionen sind u.a. O2-Langzeittherapie, intratracheale Beatmung und als ultima ratio die Lungentransplantation.
→ II: Pankreas: Substitution der Pankreasenzyme vor den Mahlzeiten bei exokriner Pankreasinsuffizienz. Auch muss ein Ausgleich der fehlenden Vitamine erfolgen. Bei Darmobstruktion sind osmotische Abführmittel wie z.B. Laktulose oder Einläufe indiziert.
→ III: Gentechnologisch wird insbesondere in England und den USA eine Übertragung für die Expression von CFTR mittels adenoviraler Vektoren oder DNA-Liposomen versucht.