- Details
- Kategorie: Dünndarm
- Zugriffe: 12479
→ Definition: Bei der Peritonealkarzinose handelt es sich um eine Besiedlung des Peritoneums mit malignen Tumorzellen. Die Tumorausbreitung erfolgt meist per continuitatem, seltener durch Tumorverschleppung im Rahmen einer operativen Tumorresektion
→ Epidemiologie: In 80% der Fälle sind Frauen im Rahmen eines Ovarialkarzinom betroffen. In 20-50% tritt sie kombiniert mit Metastasen in anderen Organen auf.
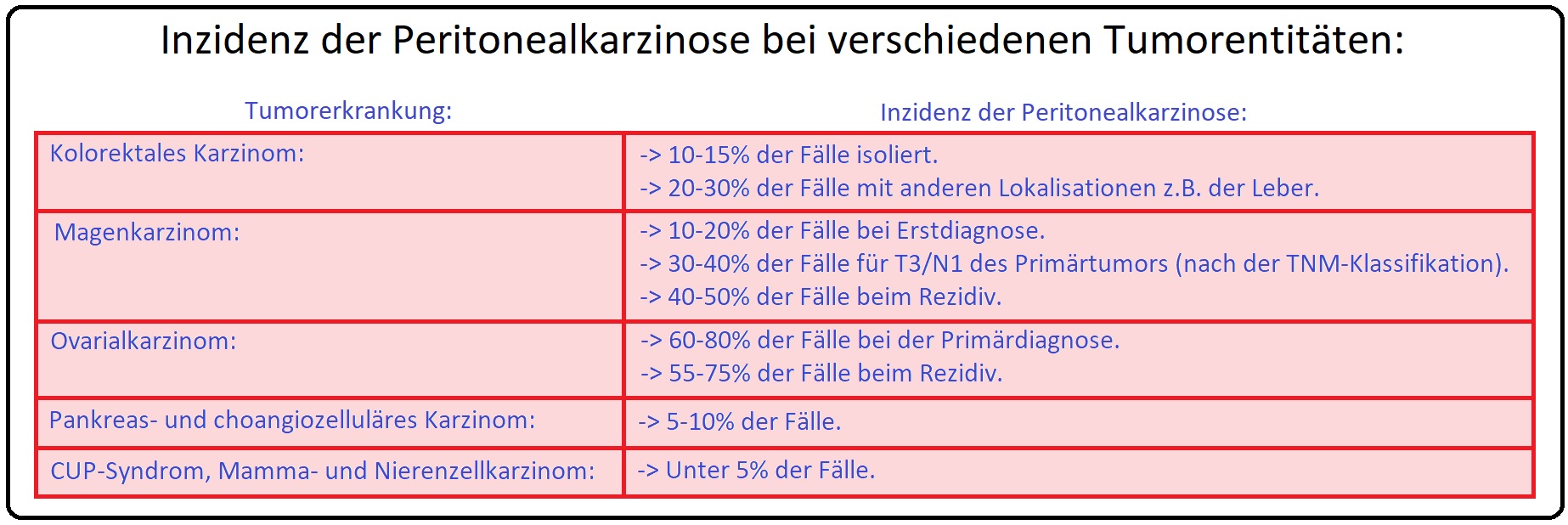
→ Ätiologie:
→ I: Durch die rasche Tumorzellproliferation und die unzureichende lymphatische Drainage gelangen einzelne Zellen intraperitoneal und werden über die Peritonealflüssigkeit verteilt.
→ II: Der daraus resultierende direkte Zell-Zell-Kontakt führt über Adhäsionsmoleküle (z.B. CD44) zur Bindung mit Mesothelzellen und konsekutivem, infiltrativem Wachstum.
→ III: Auch ist eine direkte Bindung an die extrazelluläre Matrix und Strukturen wie das Omentum majus möglich.
→ Tumorlokalisation: Mögliche Primärtumoren für die Entwicklung einer Peritonealkarzinose sind u.a. das:
→ I: Pankreaskarzinom,
→ II: Magen-Ca,
→ III: Kolorektale Karzinom sowie das
→ IV: Ovarial-Ca.
→ V: Seltenere Tumoren sind das Leber-Ca, cholangiozelluläres Karzinom, Tuben- und Uterus-Ca.
→ Klinik: Die Peritonealkarzinose ist klinisch lange Zeit inapparent und unspezifisch;
→ I: Mögliche klinische Symptome sind u.a.:
→ 1) Gewichtsabnahme,
→ 2) Verdauungsstörungen und Inappetenz,
→ 3) Verschlechterung des Allgemeinzustands,
→ 4) Ausbildung einer B-Symptomatik.
→ II: Die Manifestation eines (zumeist hämorrhagischen) Azites mit konsekutiver Zunahme des Bauchumfangs spricht für eine fortgeschrittene Tumorstreuung.
→ III: Weitere Symptome: Sind abhängig vom Tumorbefall (Symptome und Komplikationen des Primärtumor) und beinhalten u.a. Harnstau, Cholestase und mechanischer Ileus etc.
→ Diagnose:
→ I: Wichtig für die Therapie ist die umfassende Diagnose mit:
→ 1) Erhebung der Anamnese, Voroperationen, Begleiterkrankungen.
→ 2) Bestimmung der spezifischen Tumormarker:
→ A) Magenkarzinom: CA19-9, CEA, CA72-4,
→ B) Kolorektales Karzinom mit CEA und CA19-9,
→ C) Pankreaskarzinom: CA19-9,
→ D) Ovarialkarzinom: CA125, CA72-4, AFP, etc.
→ II: Sonographie: Zur Diagnosestellung, Nachweis eines Aszites und zur Aszitespunktion.

→ Klinisch-relevant: Insbesondere der diagnostische Nachweis von
→ A) Cholesterin > 45 mg/dl,
→ B) Fibronectin > 100mg/dl und
→ C) Das Vorhandensein von Tumorzellen im Aszites spricht für das Bestehen einer Peritonealkarzinose.
→ III: CT: Von Thorax, Abdomen und Becken mit konsekutiver intravenöser, oraler oder rektaler Kontrastierung zur weiteren Diagnostik des Tumorstaging, Metastasensuche, etc.

→ Differenzialdiagnose: Von der Peritonealkarzinose müssen insbesondere nachfolgende Erkrankungen abgegrenzt werden; hierzu zählen:
→ 1) Aszites anderer Genese
→ 2) Strahlenenteritis,
→ 3) Peritonitis tuberculosa.
→ Therapie:
→ I: Eine rein palliative Behandlung stellt die wiederholte Aszites-Punktion dar.
→ II: Systemische Chemotherapie:
→ 1) Die alleinige Chemotherapie verbessert die Prognose nicht, sondern wird primär nur als Kombinationsbehandlung mit einer operativen Therapie eingesetzt.
→ 2) Zu den Zytostatika gehören u.a. 5-Fluorouracil, Cisplatin, aber auch neuere Medikamente wie der Topoisomerase-Hemmer Eloxatin.
→ 3) Eine intraperitoneale Chemotherapie kann neoadjuvant zum Tumor-Downstaging appliziert werden.
→ III: Operative Therapie: Eine alleinige operative Therapie dient ausschließlich zur Palliativ-Behandlung z.B. bei Stenosierung oder Perforation. Ansonsten wird sie (in speziellen Zentren) nur in Kombination mit einer Chemotherapie zur Reduktion der Tumorgröße eingesetzt.
→ Prognose: Die Therapie der Peritonealkarzinose gestaltet sich bis heute als sehr schwierig und geht mit einer schlechten Prognose einher. Ohne Therapie liegt die mittlere Überlebenschance bei 5-6 Monaten.
- Details
- Kategorie: Dünndarm
- Zugriffe: 12656
→ Definition: Beim (Dünndarm)-Karzinoid handelt es sich um einen von den enterochromaffinen Zellen des APUD-Systems (= amine-precusor-uptake-and-decarboxylation) ausgehender epithelialer Tumor. Das Karzinoid stellt einen niedermalignen Tumor dar und produziert sowohl biogene Amine wie Serotonin, Histamin und Noradrenalin, als auch neuroendokrine Peptide (z.B. vasoaktive Substanzen wie Substanz P, Enteroglykagon,Bradykinin und Prostaglandin etc.).

→ Klinisch-relevant: Die Tumorzellen weisen zum einen morphologische und funktionelle Übereinstimmungen mit den endokrinen Zellen auf, zum anderen exprimieren sie Antigene wie Synaptophysin und Chromogranin-A, die man auch in Nervenzellen nachweisen kann.
→ Klassifikation: Nach ihrem Sekretionsprodukt unterscheidet man bei den NET zwischen:
→ I: Vorderdarmtumoren: Beinhaltet den Bereich vom Ösophagus bis zum Treitz-Band.
→ 1) Insulinom (Hypoglykämie),
→ 2) Gastrinom (peptische Ulzerationen, Diarrhoe),
→ 3) Glukagonom (Diabetes mellitus, Exanthem),
→ 4) Somatostatinom (Diabetes mellitus, Gallensteine),
→ 5) VIPom (Wässrige Diarrhoe).
→ II: Mittel- und Enddarm-Tumoren: Mitteldarm-Tumoren sind zwischen Treitz-Band und linker Kolonflexur, Enddarm- distale der linken Kolonflexur lokalisiert.
→ 1) Terminales Ileum; Serotonin mit charakteristischer Karzinoid-Symptomatik;
→ 2) Rektum meist nicht funktionell.
→ III: Extraintestinal: In 10% entwickelt sich das Karzinoid außerhalb des GIT, dann meist im Bereich des Bronchialsystems (zentral gelegene Tumoren), aber auch im Pankreas und Ovarien.
→ Epidemiologie:
→ I: Die Inzidenz des Karzinoidsyndroms liegt bei 1-2/100000 Einwohnern pro Jahr.
→ II: Das Karzinoid manifestiert sich insbesondere zwischen dem 40.-70. Lebensjahr.
→ III: Lokalisation: Die Karzinoide des Magen-Darm-Traktes bestehen:
→ 1) Zumeist in der Appendix (bis zu 50% der Fälle, gutartig und solitär); es sind aber auch Lokalisationen
→ 2) Im terminalen Ileum (in bis zu 30% der Fälle, häufig disseminiert und metastasierend),
→ 3) Im Rektum (10% der Fälle) sowie kleine Karzinoide des Magens möglich.
→ 4) Eine extragastrointestinale Manifestation stellt insbesondere das Bronchialsystem dar.
→ Ätiologie:
→ I: Die Karzinoide entwickeln sich charakteristischerweise aus einer APUD-Hyperplasie.
→ II: Im Bereich des Dünndarms wachsen sie mit einer Größe von 3mm - 3cm diffus und neigen zur Metastasierung in Leber und Lymphknoten.
→ III: Mit der Größenzunahme des Tumors steigt auch die Gefahr der Metastasierung.
→ IV: Hierdurch gelangen dann vasoaktive Substanzen wie Serotonin unter Umgehung des hepatischen Kreislaufes (die Leber inaktiviert Serotonin) in die systemische Zirkulation und lösen das klassische Karzinoid-Syndrom aus.
→ V: Karzinoide des Dickdarms produzieren kein Serotonin (5-HTA), sodass in der Folge keine Symptomatik besteht.
→ Pathophysiologie: Beim intestinalen Karzinoidtumor werden vasoaktive Mediatoren produziert, die bei fehlender hepatischer Metastasierung durch die verschiedenen Leberenzyme, insbesondere die hepatische Monoaminooxidase (Serotonin), inaktivert bzw. abgebaut werden. Erst bei Entwicklung von signifikanten Lebermetastasen verursacht der Tumor regelmäßig klinische Symptome (Aunahme bildet das Bronchialkarzinoid bei dem schon zu Krankheitsbeginn die charakteristische Symptomatik besteht). Bei manchen Patienten sind spezifische Triggermechanismen wie u.a. Alkoholzufuhr, psychischer Stress, aber auch bestimmte Nahrungsmittel oder die Palpation der Leber nachweisbar.
→ Klassifikation: Nach der WHO werden die neuroendokrinen Tumoren unterteilt in:
→ I: G1: Gut differenzierter neuroendokriner Tumor.
→ II: G2: Mäßig differenzierter neuroendokriner Tumor.
→ III: G3: Schlecht differenzierter neuroendokriner Tumor.
→ IV: G4: Gemischtes adeno-neuroendokrines Karzinom.
→ Klinik: Das Karzinoid-Syndrom ist nicht selten asymptomatisch; ansonsten manifestieren sich nachfolgende klinische Symptome:
→ I: Uncharakteristische abdominale Schmerzen z.T. kolikartig aber auch diffus mit Zeichen eines Subileus infolge lokaler Stenosierung.
→ II: Durch Sekretion von u.a. Serotonin, Kallikrein oder Bradykinin entwickelt sich in 7% der Fälle das klassische Karzinoid-Syndrom (= Cassidy-Scholte-Syndrom) und ist Ausdruck einer fortgeschrittenen Metastasierung, mit:
→ 1) Flush als Kardinalssymptom mit Hautrötung im Bereich des Gesichtes, Nackens und des oberen Thoraxbereiches und einer subjektiv unangenehm empfundenen Hitzewallung,
→ 2) Diarrhoe z.T. explosionsartige Durchfälle,
→ 3) Asthma bronchiale bzw. Asthmaanfällen infolge einer Bronchokonstriktion.
→ 4) Paroxysmale Tachykardien.
→ 5) Hedinger-Syndrom: Hierbei handelt es sich um eine (rechtsseitige) Endokardfibrose, gerade der Trikuspidalklappe mit konsekutiver Trikuspidalklappeninsuffizienz/-stenose und Rechtsherzinsuffizienz infolge der permanent erhöhten Serotoninkonzentration; seltener kann auch die Pulmonalklappe (-insuffizienz/-stenose) betroffen sein.
→ 6) Weitere Symptome: Sind u.a. Teleangiektasien (25%), Pellagra-ähnliche Dermatosen, Bronchokonstriktion (10%), Gelenkbschwerden und Arthritis, Gewichtsverlust sowie psychische Störungen (5-10%).

→ Diagnose:
→ I: Anamnese/klinische Untersuchung: In der Eigenanamnese lassen sich Symptome wie Flush, Diarrhoe, paroxysmale Tachykardien eruieren.
→ II: Labor:
→ 1) Bestimmung des Serum-Serotoninspiegels sowie der 5-Hydroxyindolessigsäure (= Abbauprodukt des Serotonins) im 24h-Sammelurin (Werte über 25mg/24h sind fast beweisend).
→ Klinisch-relevant: Medikamente wie Antihistaminika sowie zahlreiche serotoninreiche Lebensmittel wie Käse, Walnüsse, Bananen, Ananas und Tomaten sollten vor der Untersuchung abgesetzt bzw. vermieden werden.
→ 2) Ein sensitiver Tumormarker des Karzinoids ist das Chromogranin A.
→ III: Bildgebende Verfahren:
→ 1) Dient der Tumorsuche.
→ 2) Sonographie und CT zur Darstellung des Tumors bzw. seiner Lebermetastasen.
→ 3) Angiographisch erscheint das Karzinoid gefäßreich.
→ 4) Besteht ein Bronchialkarzinoid ist eine zusätzliche Bronchoskopie indiziert.
→ 5) Evtl. ist eine Somatostatin-Rezeptor-Szintigraphie anzusetzen.
→ Differenzialdiagnose: Hiervon abzugrenzen ist das (Abb.: Gastroenteropankreatische Neoplasien):
→ I: Gastrinom des Duodenums,
→ II: Phäochromozytom,
→ III: Systemische Mastozytose,
→ IV: Non-Hodgkin-Lymphom, etc.
→ Therapie: Die Therapie des Karzinoidsyndroms ist fast ausschließlich palliativ und symptomatisch, weil beim Auftreten der typischen Symptomen bereits eine ausgedehnte Metastasierung besteht.
→ I: Allgemeinmaßnahmen: Auslösende Triggerfaktoren sollten gemieden werden; hierzu zählen insbesondere:
→ 1) Alkohol,
→ 2) Stark gewürzte Nahrungsmittel und
→ 3) Körperliche Anstrengung.
→ II: Operative Therapie:
→ 1) Mittel der Wahl ist die operative Resektion des Primärtumors sowie der lokalen Lymphknoten.
→ 2) Beim Karzinoid der Appendix (< 1cm) ist eine alleinige Appendektomie ausreichend.
→ 3) Bei Tumoren an der Appendixbasis mit nachgewiesenen LK-Mestastasen sollte eine Hemikolektomie rechts erfolgen.
→ 4) Lebermetastasen sollten reseziert oder chemoembolisiert werden.
→ Klinisch-relevant: Eine Karzinoidkrise kann jeder Zeit intraoperativ auftreten, manifestiert sich jedoch besonders häufig bei Manipulation im Tumorgebiet. Durch intravenöse Bolus-Applikation von Octreotid (25-50 µg) oder kontinuierlicher Infusion von 100 µg Octreotid und weiterer Serotoninantagonisten (z.B. Ondansetron 8 mg) kann die klinische Symptomatik infolge einer überschießenden Serotoninausschüttung abgemildert bzw. reduziert werden. Aprotinin, ein Kallikrein-Inhibitor, kann zur Verhinderung von Blutungskomplikationen eingesetzt werden.
→ III: Medikamentöse Therapie:
→ 1) Bei Inoperabilität und ausgedehnter Metastasierung werden Somatostatin-Analoga wie z.B. Octreotid appliziert.
→ 2) Sandostatin 3x tgl. s.c. oder i.m. als Monatsdepot.
→ 3) Somatostatin Analoga hemmen die Hormonsekretion sowie das weitere Wachstum des Tumors, sodass die klinische Symptomatik weitestgehend unterbunden werden kann.
→ 4) Die Flush-Anfälle können zusätzlich mit z.B. Dibenzyran 10-30 mg behandelt werden.

→ Prognose: Bei lokalisiertem Karzinoidsyndrom liegt die 5-Jahresüberlebenschance bei 65%, bei Inoperabilität ist trotz des langsamen Wachstums die Prognose eher ungünstig und weist eine 5-Jahresüberlebenschance von nur 30-35% auf.