→ Definition: Bei der IgA-Nephropathie handelt es sich um eine (proliferative) Glomerulonephritis, bei der es zur Ablagerung von Immunglobulinen der Klasse A1 in den mesangialen Zellen des Glomerulus kommt. Zusätzlich manifestiert sich eine Proliferation der Mesangialzellen und Matrixvermehrung.
→ Epidemiologie:
→ I: Die IgA-Nephropathie ist weltweit die häufigste Form der idiopathischen Glomerulonephritis. Jährlich ist mit 20-40 Neuerkrankungen/1 Million Einwohnern zu rechnen.
→ II: Der Manifestationsgipfel liegt in der 2. bis 4. Lebensdekade (kann aber in jedem Lebensalter auftreten), wobei Männer doppelt so häufig wie Frauen betroffen sind.
→ Ätiologie:
→ I: Der Morbus Berger tritt zumeist idiopathisch auf; hierbei wird eine gestörte Produktion von polyklonalen IgA-1 Antikörpern im Knochenmark und den lymphatischen Geweben und oder eine strukturelle Störung des IgA-Moleküls durch unzureichende Glykolisierung wird diskutiert.
→ II: Der Morbus Berger ist aber auch mit weiteren Erkrankungen (z.B. Pupura Schoenlein-Henoch,) assoziiert. Der zugrunde liegende Pathomechanismus ist jedoch bis heute nicht genau geklärt. Wichtige Vorerkrankungen sind z.B. die
→ 1) Leberzirrhose, Hepatitis B und die primär biliäre Zirrhose,
→ 2) Chronisch entzündliche Darmerkrankungen wie Morbus Crohn und Colitis ulcerosa, aber auch die glutensensitive Enteropathie.
→ 3) HLA-B27-Arthritiden, selten infolge einer Sarkoidose, Hämosiderose, etc.
→ III: Eine genetische Disposition aufgrund einer positiven Familienanamnese zeigt sich in 30% der Fälle, insbesondere bei erhöhter HLA-DR4 Frequenz.

→ Pathogenese:
→ I: Beim Morbus Berger wurde eine Mutation auf dem (6q22-23) Gen lokalisiert, das wahrscheinlich mit der Erkrankung in direktem Zusammenhang steht.
→ II: Bei den in den mensangial abgelagerten Immunglobulinen handelt es sich um polymere IgA der Unterklasse A1, die vorwiegend im Knochenmark (B-Lymphozyten) produziert werden. Das Immunglobulin ist an seinem Serin- bzw. Threonin-Rest mit Kohlenhydraten atypisch glykosyliert und eine weitere glykosidische Bindung mit Galaktose ist aufgrund eines ß1-3-Galaktosyltransferase-Defektes nicht möglich. Folge ist Bildung von IgA-Polymeren, die die Interaktion mit IgA-Rezeptoren (CD89) auf myeloiden Zellen und Transferrin-Rezeptoren (CD71) auf Mesangialzellen stören. Angenommen wird eine vermehrte Freisetzung dieser Rezeptoren, die konsekutiv zu zirkulierenden Komplexen führen.
→ III: Im Anschluss an die mesangiale Ablagerung, die einen Entzündungsreiz darstellt, kommt es durch Zytokine wie das Interleucin-6, der Plättchen-Wachstumsfaktor PDGF, Tumornekrosefaktor-Alpha und Beta, die als Mediatoren wirken, zur masangialen Proliferation und Einwanderung von Entzündungszellen.
→ Klinik: Die klinische Symptomatik und der Krankheitsverlauf der IgA-Nephropathie sind durch das Auftreten verschiedener glomerulärer Syndrome sehr variabel:
→ I: Oft zeigt sich zu Beginn der Erkrankung eine asymptomatische, schmerzlose Makro- bzw. Mikrohämaturie (mit/ohne Proteinurie), die intermittieren oder nach einigen Tagen sistieren kann.
→ II: Weitere Symptome: Insbesondere bei rapidem Krankheitsverlauf manifestieren sich Symptome wie Einschränkung der glomerulären Filtrationsrate, arterielle Hypertonie, nephrotisches Syndrom bis hin zu einem akuten Nierenversagen.
→ III: Eine chronische Niereninsuffizienz manifestiert sich in bis zu 30% der Fälle im weiteren Krankheitsverlauf. (10% der Betroffenen sind nach 10 Jahren dialysepflichtig).
→ IV: Nicht selten ist eine unspezifische Infektion der oberen Atemwege eruierbar.
→ Diagnose:
→ I: Anamnese/Klinische Untersuchung:
→ 1) Mit Abklärung wiederholter Makrohämaturie, vorausgegangener Infekte der oberen Atemwege (1-3 Tagen zuvor) oder Gastroenteritiden etc.
→ 2) Nachweis einer arteriellen Hypertonie.
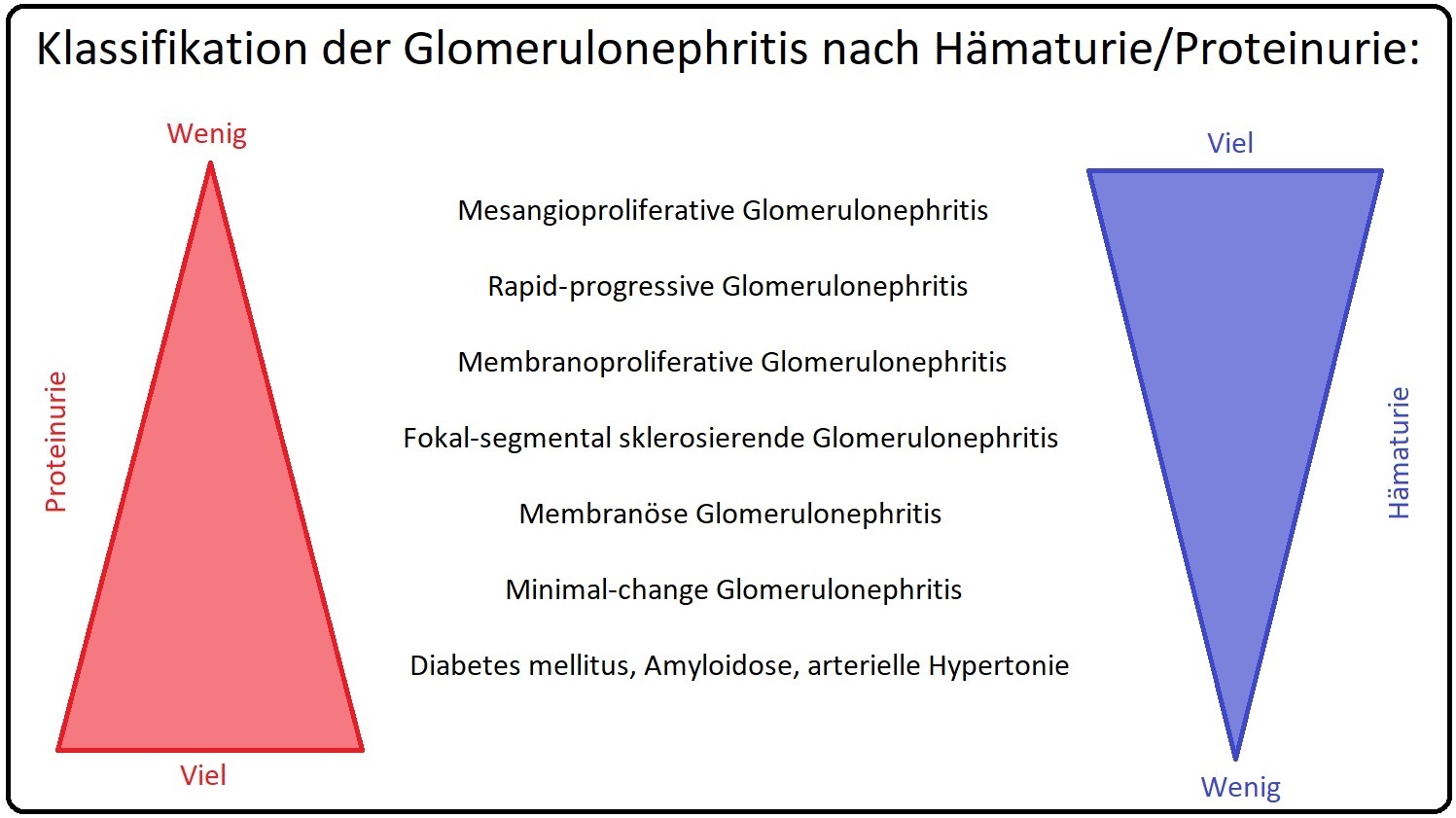
→ II: Labor:
→ 1) Erhöhte IgA-Spiegel im Serum, sowie zirkulierenden Immunkomplexen, die IgA enthalten. Ihre Konzentration korreliert mit der Krankheitsaktivität.
→ 2) Urin: Mit Nachweis einer Hämaturie sowie Erythrozytenzylindern und dysmorphen Erythrozyten; zudem besteht nicht selten eine unselektive Proteinurie (< 3g/d). Diese einfache Untersuchung kann vielen Patienten mit IgA-Nephropathie die häufig durchgeführten Untersuchungen i.v. Urographie und Zystoskopie ersparen.
→ 3) Nachfolgende Prädiktoren, nämlich Verminderung der glomerulären Filtrationsrate, Proteinurie > 1g/d sowie renale Hypertonie, weisen auf eine ungünstige renale Langzeitprognose hin.
→ III: Nierenbiopsie: Die eindeutige Diagnose der IgA-Nephropathie kann jedoch nur durch eine immunhistochemische Untersuchung des Nierenbiopsats gestellt werden. Typisch sind IgA und C3-Ablagerungen überwiegend im Bereich des Mesangiums und mithilfe der Immunfluoreszenz in allen Glomeruli nachweisbar.

→ IV: Klinischer-Prognose-Index: Er beinhaltet 4 klinische Parameter und dient der Erfassung einer möglichen Nierenfunktionsverschlechterung.
→ 1) Serumkreatinin-Konzentration > 1,4mg/dl = 2 Punkte.
→ 2) Proteinurie > 1g/d = 1 Punkt.
→ 3) Arterielle Hypertonie = 1 Punkt.
→ 4) Alter > 30. Lebensjahr = 1 Punkt.
→ 5) Beurteilung: Die renale 10-Jahres-Überlebensate beträgt bei 0-2 Punkten 90% und bei 3-5 Punkten nur noch 35%.
→ Differenzialdiagnose: Vom Morbus Berger müssen insbesondere nachfolgende Erkrankungen abgegrenzt werden; hierzu zählen:
→ I: Membranoproliferative Glomerulonephritis,
→ II: Poststreptokokken-Glomerulonephritis (2-3 Wochen nach einer Infektion mit Streptokokken).

→ III: Pupura Schoenlein-Henoch und
→ IV: Weitere Differenzialdiagnose: Sind insbesondere:
→ 1) Patienten mit Leberzirrhose und Zöliakie.
→ 2) Analgetikanephropathie und Nephrolithiasis.
→ 3) Das Syndrom der dünnen Basalmembran, aber auch
→ 4) Bei Hauterkrankungen wie Dermatitis herpetiformis und Psoriasis vulgaris.
→ Therapie:
→ I: Persistiert lediglich einer isolierte Hämaturie (ohne Proteinurie und arterieller Hypertonie) ist eine medikamentöse Therapie nicht indiziert und es werden jährliche Kontrolluntersuchungen empfohlen.
→ II: Medikamentöse Therapie:
→ 1) Besteht jedoch zusätzlich eine Proteinurie (> 0,5g/d) oder einer arterielle Hypertonie (> 130/80mmHg) oder eine eingeschränkte glomeruläre Filtrationsrate wird ein ACE-Hemmer oder ein AT-1-Blocker über einen Zeitraum von 3-6 Monaten appliziert.
→ 2) Persistiert die Proteinurie trotz 6-monatiger antihypertensiver Therapie bzw. überschreitet sie > 1g/d oder kommt es zur weiteren Abnahme der glomerulären Filtrationsrate sollte eine Therapie mit einem Glukokortikoid nach dem Pozzi-Schema über einen Zeitraum von 6 Monaten versucht werden.

→ III: Bei einer akuten und raschen Reduktion der glomerulären Filtrationsrate (in Form einer rapid-progressiven GN) ist eine immunsuppressive Behandlung mit einem Glukokortikoid und Cyclophosphamid obligat.

→ Prognose:
→ I: Die IgA-Nephropathie verläuft nicht selten gutartig (in 10% der Fälle kommt es zu einer spontanen Remission), jedoch entwickeln 20-30% der Erkrankten eine terminale Niereninsuffizienz.
→ II: Insbesondere die Proteinurie stellt einen wichtigen Prognosefaktor dar. Eine Proteinurie > 3g/d weist über die Jahre einen signifikanten Nierenfunktionsverlust auf (Verlust der GFR von > 9ml/min pro Jahr).
→ III: Weitere ungünstige Faktoren für die Langzeitprognose sind u.a.:
→ 1) Höheres Lebensalter.
→ 2) Männliches Geschlecht.
→ 3) Hohe arterielle Blutdruckwerte (arterielle Hypertonie).
→ 4) Eine glomeruläre Filtrationsrate (GFR) von < 60ml/min und
→ 5) Das histologische Bild einer glomerulären oder interstitiellen Sklerosierung.