- Details
- Kategorie: Störungen des Knochenstoffwechsels / metabolische Osteopathien
- Zugriffe: 9343
→ Definition:
→ I: Osteomalazie: Hierbei handelt es sich um eine generalisierte (systemische) Knochenstoffwechselstörung des primär gesunden Knochens mit unzureichender Mineralisation des neugebildeten Osteoids (= weiche noch nicht mineralisierte, von den Osteoblasten gebildete Grundsubstanz bzw. Matrix) von Kompakta und Spongiosa infolge eines verminderten Kalzium- und Phosphatangebotes unterschiedlicher Genese. Hierdurch wird der Knochen weich und verbiegbar.
→ II: Rachitis: Bei der Rachitis des Kindes besteht eine gestörte Mineralisation des Skeletts und Desorganisation der metaphysären Wachstumsfuge beim primär geschädigten Knochen (= Störung der normalen Knochenentwicklung).
→ Klinisch-relevant:
→ A) Wird die Knochenstoffwechselstörung zwischen dem 3. Lebensmonat und 3. Lebensjahr diagnostiziert, spricht man von Rachitis,
→ B) Eine Manifestation zwischen dem 3. Lebensjahr und der Pubertät wird "Spätrachitis" genannt.
→ Epidemiologie: Das Vollbild der Osteomalazie ist heute in Deutschland nur noch selten anzutreffen. Besonders betroffen sind:
→ I: Ältere Patienten mit fehlender Sonnenexposition,
→ II: Ein Osteomalazie-ähnliches Krankheitsbild wird z.B. durch die Einnahme des Antiepileptikums, Hydantoin, erreicht, da es mit dem Vitamin-D-Stoffwechsel interferiert.
→ Physiologie:
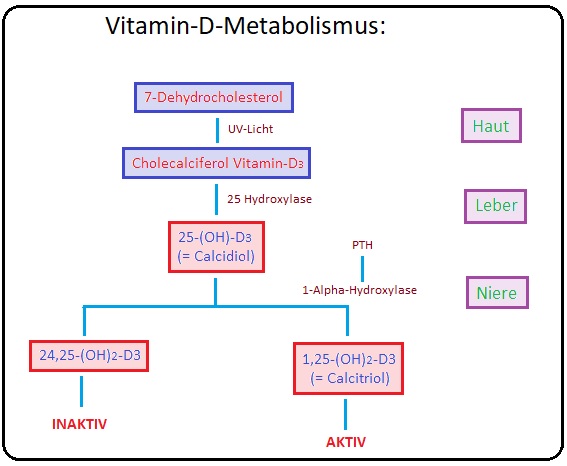
→ I: Die Vitamin-D-Synthese beginnt in der Haut durch Bildung von Cholecalciferol aus 7-Dehydrocholesterol.
→ II: Anschließend wird es in der Leber zu 25-Hydroxycholecalciferol (= Cholecacifediol) und schließlich in der Niere in 1-25-Dihydroxycholecalciferol (= aktives Vitamin-D-Hormon). hydroxyliert.
→ III: Parathormon oder eine Hypokalzämie fördern die Bildung des aktiven Vitamin-D-Hormons mit konsekutiv vermehrter enteraler Kalziumresorption und gesteigerter Osteogenese.
→ Ätiologie: Der Osteomalazie liegen v.a. Störungen des Vitamin-D-Stoffwechsels oder Phosphatstoffwechsels zugrunde (eine seltenere Ursache ist z.B. die Bisphosphonat-Behandlung).
→ I: Vitamin-D-Mangel:
→ 1) Zu geringe exogene Vitamin-D-Zufuhr (Mangelernährung etc.).
→ 2) Unzureichende Sonnenexposition,
→ 3) Gastrointestinale Störungen: Wie Malassimilationssyndrom infolge z.B. einer Zöliakie, eines Morbus Whipple oder Morbus Crohn, aber auch Zustand nach Gastrektomie, Billroth-II-Operation, Dünndarmresektion, etc.
→ 4) Erhöhter Vit-D-Metabolismus: Bei der Einnahme von Antiepileptika (Hydantoin), Rifampizin und Glutethimid.
→ II: Störungen des Vit-D-Stoffwechsels:
→ 1) Leber: Verminderte 25-Hydroxylierung in der Leber bei Leberparenchym-Erkrankungen wie Leberzirrhose, primär-biliärer-Zirrhose oder chronisch-aktiver Hepatitis. Folge ist eine verminderte Bildung von 25-Hydroxycholecalciferol.
→ 2) Niere: Zumeist infolge chronischer Niereninsuffizienz, selten aufgrund einer Vitamin-D-resistente Rachitis. Es werden 2 Typen unterschieden:
→ A) VDDR-Typ 1: Es besteht eine Mutation der 1-Alpha-Hydroxylase.
→ B) VDDR-Typ 2: Mit einer genetisch bedingten Störung des intrazellulären Vitamin-D-Rezeptors.
Weitere renale Ursachen sind u.a. chronische Pyelonephritis, interstitielle Nephritis, Glomerulonephritis, polyzystische Nieren, etc.
→ III: Vitamin-D-unabhängige Osteomalazie: Bei renal-tubulären Nierenfunktionsstörungen wie u.a.:
→ 1) Bei gesteigertem renalem Phosphatverlust im Bereich des proximaler Tubulus, infolge eines Phosphatdiabetes (hereditär X-chromosomal vererbt), idiopathisch bei Erwachsenen, onkogen bedingt bei mesenchymalen Tumoren, die den Fibroblasten-Growth-Faktor 23 sezernieren, der wiederum die Wideraufnahme des Phosphat über den Phosphattransporter in der Niere hemmt.
→ 2) Phosphaturische-glukosurische-aminoazidurische Form des De-Toni-Debre-Fanconi-Syndroms.
→ 3) Renal-tubuläre Azidose.
→ 4) Hypophosphatasie: Hierbei handelt es sich um eine autosomal-rezessiv vererbte Erkrankung, bei der die Synthese der alkalischen Phosphatase in den Osteoblasten deutlich vermindert ist.

→ Klinik: Charakteristische klinische Beschwerden sind u.a.:
→ I: Osteomalazie:
→ 1) Diffuse generalisierte Knochenschmerzen, vor allem auch im Becken- und Hüftbereich, aber auch Schmerzen im Adduktorenbereich sowie Fersenschmerzen.
→ 2) Knochendeformierungen mit u.a. Entwicklung einer Coxa vara (Femurknochen), eines Genu varum (Tibia) sowie einer Wirbelsäulenkyphose.
→ 3) Nicht selten manifestieren sich Ermüdungsfrakturen. Diese findet man insbesondere im Bereich des Os ischii und Os pubis mit den für die Patienten mit Osteomalazie charakteristischen Leistenschmerzen.
→ 4) Häufig bildet sich eine Schwäche im Bereich der proximalen Muskelgruppen aus z.B. Myopathie der Glutealmuskulatur (aufgrund einer intrazellulären Phosphatverarmung) mit konsekutivem Watschelgang.
→ 5) Infolge der Hypokalzämie können sich evtl. tetanische Zustände entwickeln.
→ II: Rachitis:
→ 1) Fehlender Fontanellenschluss mit Abflachung des Hinterkopfes (Kraniotabes),
→ 2) Aufreibungen im Bereich der Übergangszone zwischen Rippenknorpel und -knochen (rachitischer Rosenkranz).
→ 3) Deformierungen des gesamten Thorax (= Glockenthorax) und evtl. auch des Beckens.
→ 4) Gestörte Zahnbildung.
→ Diagnose:
→ I: Anamnese (Grunderkrankung) und klinische Untersuchung geben erste Hinweise.
→ II: Labor:
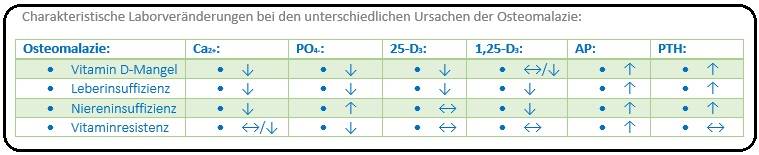
→ 1) Vit-D-abhängiger Osteomalazie:
→ A) Hypokalzämie, Hypophosphatämie (z.T. infolge eines sekundären Hypoparathyreoidismus), Erhöhung der alkalischen Phosphatase und des Parathormons.
→ B) Beim Malassimilations-Syndrom (Malabsorption) besteht ein Mangel an 25-OH-D3,
→ C) Bei der Niereninsuffizienz manifestiert sich ein 1-25-(OH)2-D3 Mangel sowie eine Hyperphosphatämie.
→ 2) Vitamin-D-unabhängige-Osteomalazie: Im Zusammenhang mit einer renalen Tubulopathie ( z.B. Fanconi-Syndrom, renal-tubuläre Azidose) lassen sich folgende laborchemische Veränderungen nachweisen: Kalzium im Normbereich (2,1-2,5mmol/l bzw. ionisiertes Ca2+ 1,15-1,3mmol/l), Hypophosphatämie bei erhöhter alkalischer Phosphatase und normalem Parathormon.
→ III: Röntgen:
→ 1) Typisch bei der Osteomalazie ist eine generalisierte Transparenzerhöhung des Knochens mit verwaschenen, unscharf begrenzten Spongiosastrukturen (= Mattglasphänomen),
→ 2) Im fortgeschrittenen Stadium erscheint die Compacta unscharf begrenzt und ausgedünnt.
→ 3) Charakteristikum sind die „ Looser-Umbauzonen“ (Pseudofrakturen; DD: sind auch bei der Osteoporose nachweibar). Diese stellen sich initial als schlecht abgrenzbare orthogonal zur Längsachse des Knochens verlaufende Aufhellungslinien in der Kortikalis (= belastungsinduzierte kortikale Frakturen) dar. Prädilektionsstellen sind insbesondere Femurhals, Femurschaft, Rippen, Scham- und Sitzbein, sowie die langen Röhrenknochen.
→ Klinisch-relevant: Treten diese Pseudofrakturen (= spaltförmige Aufhellung = Kontinuitätsunterbrechungen) beidseits an einem Knochen auf spricht man vom Milkman-Syndrom.
→ 4) Weitere radiologische Auffälligkeiten sind Skelettveränderungen wie Kyphoskoliose, bikonkave Fischwirbel und nicht zuletzt deutliche Verbiegungen der proximalen Extremitätenknochen.
→ 5) Rachitis: Hierbei zeigt sich radiologisch:
→ A) Axiale Verbreiterung der Epiphyse (= Becherung).
→ B) Pinselartige unregelmäßige Aufhellungslinien am Übergang von Metaphyse zu Diaphyse.
→ C) Knochendeformierungen wie Glockenthorax, kartenherzartiges Becken und rachitischer Rosenkranz.
→ IV: Nur selten ist eine Knochenbiopsie (Beckenkamm) zur Diagnosestellung indiziert.
→ Therapie:
→ I: Bei der Vitamin-D-abhängigen Osteomalazie ist eine Vitamin-D3-Substitution mit einer Dosierung von 10000IE/d per os über 3 Wochen indiziert. Im weiteren Behandlungsverlauf wird die Dosis auf 1000IE/d reduziert.
→ II: Bei Leberfunktionsstörungen oder einer LZ-Therapie mit Antiepileptika kann 25-OH-Cholecalciferol, bei Niereninsuffizienz 1-25-(OH)2-Cholecaciferol verabreicht werden.
→ III: Beim Malassimilationssyndrom muss eine parenterale Applikation der fettlöslichen Vitamine (EDKA) erfolgen. Die Dosis richtet sich nach der Kalziumkonzentration.
→ IV: Im Kindesalter sollte präventiv eine Vitamin-D-Prophylaxe eingeleitet werden.
→ V: Evtl. ist die Behandlung der renalen Osteopathie indiziert.
→ Klinisch-relevant: Der Therapieerfolg wird durch die Abnahme der alkalischen Phosphatase (nach Wochen bis Monaten) laborchemisch gesichert.
- Details
- Kategorie: Störungen des Knochenstoffwechsels / metabolische Osteopathien
- Zugriffe: 7675
→ Definition: Beim Morbus Paget handelt es sich um eine lokalisierte mono- oder polyostotische, progressive Skeletterkrankung aufgrund von pathologisch vermehrten Knochenumbauprozessen. Folge ist die Ausbildung eines mechanisch-minderwertigen Knochens (= Geflechtknochen) mit erhöhter Frakturgefahr, Schmerzen und Deformität.
→ Epidemiologie:
→ I: Die Osteodystrophia deformans stellt nach der Osteoporose die 2. häufigste Knochenerkrankung (die Prävalenz liegt in Westeuropa bei 1-3%) dar; es sind gerade Menschen aus dem anglikanischen Raum betroffen.
→ II: Der Manifestationsgipfel liegt im höheren Lebensalter, jenseits des 40. Lebensjahrs, wobei Männer marginal häufiger als Frauen betroffen sind.
→ Ätiologie:
→ I: Die Ursachen sind nicht genau bekannt, jedoch werden vorangegangene Virusinfektionen diskutiert (Low-virus infection);
→ II: In 30% der Fälle findet man eine Mutation des RANK-Gens, welches eine genetische Disposition (Chromosom 18q) unterstreicht.
→ Pathogenese:
→ I: Frühphase: (= lytisches Stadium) In dieser Phase manifestiert sich eine überschießende pathologische Anzahl und Aktivität der Osteoklasten (= Riesenosteoklasten, die intranukleäre Einschlüsse) mit konsekutivem beschleunigtem Knochenabbau.
→ II: Spätphase: (= gemischtes Stadium) Es erfolgt ein kompensatorischer Knochenanbau durch Osteoblasten.
→ III: Sklerotisches Stadium: Folge ist die Bildung eines mechanisch-minderwertigen (vermindert-mineralisierten) Geflechtknochens, der verdickt, deformiert, hypervaskularisiert und nicht zuletzt begrenzt belastbar ist.
→ Lokalisation:
→ I: Der Morbus Paget kann mono-, oligo- oder polyostotisch auftreten.
→ II: Am häufigsten ist das Achsenskelett (Schädel, Wirbelsäule, insbesondere die LWS, Becken) sowie der Femur und die Tibia betroffen. Grundsätzlich können aber auch alle Knochen befallen sein-
→ Klinik:
→ I: Meist verläuft der Morbus Paget symptomlos und wird im Zuge weiterer radiologischer Untersuchungen als Zufallsbefund eruiert.
→ II: Bestehen jedoch klinische Symptome, sind diese charakteristisch:
→ 1) Langsam sich entwickelnde z.T. groteske Deformität des Knochens (z.B. Säbelscheiden-Tibia).
→ 2) Initial bestehen meist nur geringe Schmerzen, die im weiteren Krankheitsverlauf deutlich zunehmen können.
→ 3) Pathologische Frakturen durch Fehlbelastung des untermineralisierten Faserknochens.
→ 4) Bei Befall der Wirbelsäule können sich Sinterung (= allmähliche Höhenminderung der Knochenstruktur infolge degenerativer Prozesse) der Wirbelkörper sowie bei Befall des Wirbelbogens Einengungen der Foramina vertebralis (Spinalkanals) mit konsekutiven radikulären Schmerzen manifestieren; nur seltener bilden sich sensormotorische Ausfälle aus.
→ 5) Bei Befall des Schädels kommt es zu einer deutlichen Schädelverformung (= "Löwenhaupt") und der Kopfumfang nimmt zu; selten entwickeln sich Kopfschmerzen oder eine Schwerhörigkeit (Schallempfindungsstörung) infolge einer Miteinbeziehung der Pars petrosa ossis temporalis.
→ III: Weitere Symptome: Sind u.a. Myalgien aufgrund von Muskelfehlbelastungen, Sekundärarthrosen im Bereich der angrenzenden Gelenke, lokale Hyperthermie, etc.

→ Klinisch-relevant:
→ A) Im floriden Stadium bestehen z.T. ausgeprägte Knochenschmerzen mit lokaler Überwärmung evtl. unter Miteinbeziehung der anliegenden Gelenke.
→ B) Die betroffenen Paget-Knochen sind sehr frakturanfällig.
→ Komplikation: Wichtige z.T. schwerwiegende Komplikationen bei der Osteodystrophia deformans sind u.a.:
→ I: Sekundärarthrose aufgrund der Knochenfehlstellung sowie Frakturneigung.
→ II: Allgemein Kompressionsyndrome wie z.B. Wurzelkompressionssyndrom bis hin zu Paraplegie bei Befall der WS, Hirnnervenkompressionssyndrom, etc.
→ III: Nierensteine bei Hyperkalzämie.
→ IV: Evtl. Herzinsuffizienz durch Volumenbelastung infolge einer vermehrten Knochendurchblutung.
→ V: In 1% der Fälle kann sich eine tumorröse Entartung (z.B. Osteosarkom) manifestieren. Klinische Hinweise hierfür sind Zunahme der Schmerzen, zunehmende Hyperthermie der betroffenen Regionen, deutlicher Anstieg der Entzündungsparameter im Blut sowie radiologische Aufhellungsareale in zuvor verdichteten Zonen. Das Osteosarkom metastasiert frühzeitig und weist eine schlechte Prognose auf.
→ Diagnose:
→ I: Anamnese/klinische Untersuchung:
→ 1) Lokale in den spezifischen Bereichen bestehende Knochenschmerzen, evtl, Deformitäten.
→ II: Labor: Trotz des 20-fach erhöhten Kalziumumsatzes sind die Serumspiegel für Kalzium und Phosphat zumeist regelrecht.
→ 1) Spezifischer Knochenmarker ist das Isoenzym der alkalischen Phosphatase, die Knochen-AP. Ihre Bestimmung dient der Diagnosestellung und stellt im Krankheitsverlauf einen Aktivitätsparameter dar.
→ 2) Je nach Ausmaß der Knochenumbauvorgänge ist die Hydroxyprolin-Ausscheidung im Urin (deutlich) gesteigert.
→ III: Bildgebende Verfahren:
→ 1) Röntgen: Das rein lytische oder rein sklerotische Stadium lässt sich radiologisch selten beobachten, vielmehr erleichtert das Nebeneinander lytischer und sklerotischer Veränderungen im gemischten Stadium die Diagnose des Morbus Paget.
→ A) 1. Phase: Frühphase mit Nachweis von Osteolysen, die gelenksnah beginnen und sich schaftwärts ausbreiten.
→ B) 2. Phase: Mischbild zwischen osteolytischen Destruktionen und osteosklerotischen Arealen.
→ Klinisch relevant: Die radiologisch nachweisbare Verdickung des betroffenen Knochens mit „ strähniger Aufblätterung" der Kompakta ist kennzeichnend für den Morbus Paget.
→ C) 3. Phase: Der betroffene Knochen nimmt an Dichte und Volumen zu und ist durch eine relative homogene Sklerosierung geprägt (= Sklerosestadium).

→ 2) MRT: Der Morbus Paget ist eine der seltenen Erkrankungen, die mit einem unauffälligem MRT-Befund einhergehen kann. Insbesondere im Knochenmark können aber auch abhängig vom Stadium reaktive Veränaderungen eruierbar sein. Das Knochenmark ist T1-gewichtet hierbei signalvermindert, T2- gewichtet signalverstärkt und nach Kontrastmittelgabe wird eine Hypervaskularisation dargestellt. In der Regel findet man nultiple steifig bis punktförmige eingelagerte Fettmarkreste (T1-geichtet hell), sodass sich ein gespenkeltes Bild ergibt. Des Weiteren ist in allen Sequenzen typischerweise als Ausdruck der Sklerose das Signal vermindert.
→ 3) Szintigraphie: In den Paget-Arealen stellt sich aufgrund des erhöhten Knochenumbaus eine ausgeprägte Aktivitätsanreicherung dar.
→ Differenzialdiagnose: Vom Morbus Paget müssen insbesondere nachfolgende Erkankungen abgegrenzt werden:
→ I: Knochenmetastasen,
→ II: Osteomyelitis,
→ III: Primärer Hyperparathyreoidismus.

→ Therapie: Wichtige Therapieziele beim Morbus Paget sind u.a. die Osteoklastenhemmung, die Schmerzreduktion und nicht zuletzt die Verhindeurng von Deformierungen.
→ I: Absolute Indikation: Knochenschmerzen, Knochendeformität, Frakturanfälligkeiten, Schädelbefall, ausgeprägte Umbauaktivität mit AP 600-800E/l und nicht zuletzt neurologische Komplikationen.
→ II: Relative Indikation: Jüngeres Alter, radiologische Progression der Erkrankung, Herzinsuffizienz infolge einer vermehrten Knochendurchblutung.
→ III: Medikamentöse Therapie: Mittel der 1. Wahl sind die Bisposphonate wie z.B.:
→ 1) Risedonat 30mg/d per os über 3-6 Monate;
→ 2) Pamidonat 30-90mg i.v. alle 4 Wochen über 3 Monate oder
→ 3) Zoledonat 5mg als Kurzinfusion 1x/Jahr.
→ 4) Calcitonin hat in der Behandlung des Morbus Paget durch Hemmung der Osteoklasten sowie der antihyperkalzämischen und analgetischen Wirkung einen festen Stellenwert.
Nach Beginn der medikamentösen Therapie sollte nach 3 Monaten die erste Serum-AP-Bestimmung erfolgen; anschließend alle 6 Monate. Bei einem erneuten Anstieg der AP über 25% des minimalsten AP-Wertes oder bei Überschreiten des Normbereiches sollte erneut mit einer medikamentösen Therapie begonnen werden.

→ IV: Weitere Maßnahmen: Sind insbesondere:
→ 1) Analgetika-Therapie wie ASS und Diclofenac zur Schmerzbehandlung.
→ 2) Krankengymnastik.
→ 3) Ausreichende Zufuhr von Kalzium und Vitamin D. Diese sollten mit einem Abstand von 2 Stunden eingenommen werden, da ansonsten die Bisphosphonate schlechter resorbiert werden.
→ Prognose: Die Prognose des Morbus Paget richtet sich im Allgemeinen nach der Ausdehnung und der Lokalisation der Knochenveränderungen. Wichtige weitere Prognosefraktoren sind u.a.:
→ I: Eine frühzeitige adäquate Behandlung, um Knochendeformitäten zu verhindern bzw. eine Beschwerdefreiheit zu erreichen.
→ II: In weniger als 1% entwickelt sich ein Paget-Sarkom.