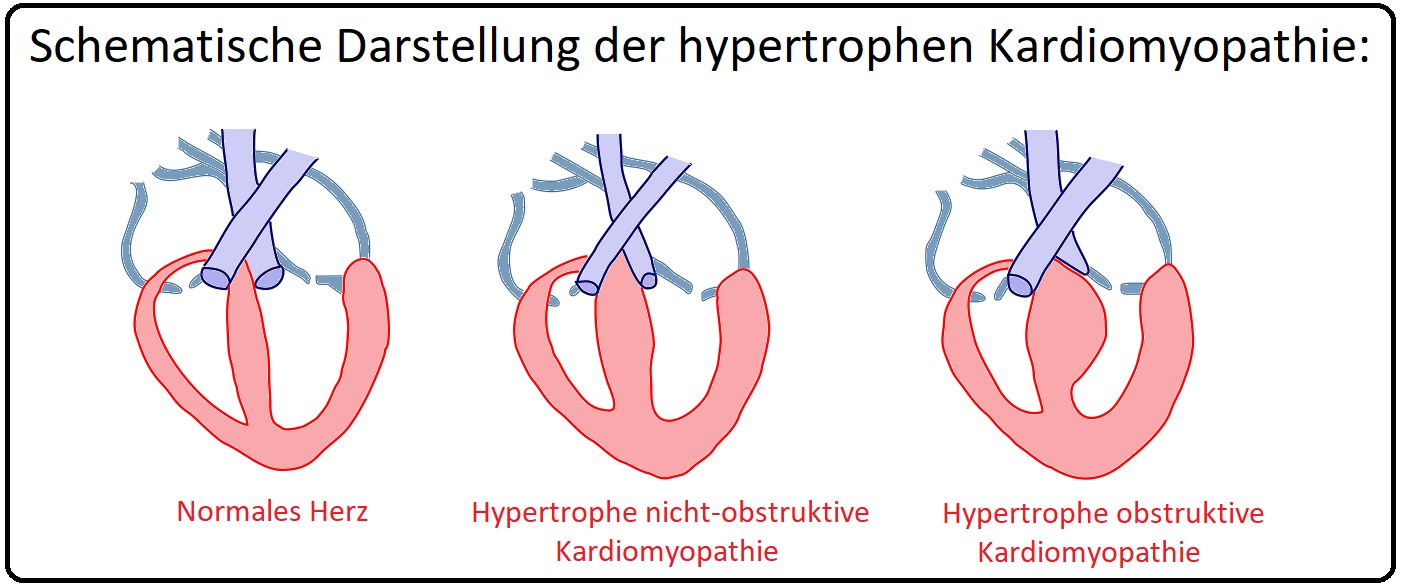
→ Definition: Die hypertrophe Kardiomypathie ist charakterisiert durch eine pathologische Hypertrophie des linken Ventrikels (gerade im septalen Bereich) ohne adäquate Druckbelastung wie z.B. arterielle Hypertonie oder Aortenklappenstenose. Kommt es während der Systole im Bereich der linken Ausflussbahn zur Obstruktion mit Steigerung des Druckgradienten spricht man von einer hypertrophen-obstruktiven-Kardiomyopathie.
→ Klassifikation: Diese Form der Kardiomyopathie wird nochmals unterteilt in eine:
→ I: Hypertrophe-Nichtobstruktive-Kardiomyopathie (HNCM): Mit 3/4 die häufigere Form;
→ II: Hypertrophe-Obstruktive-Kardiomyopathie (HOCM): Mit 1/4 die seltenere Form. Sie wird genetisch vererbt und führt oftmals zu einer asymmetrischen Hypertrophie des linksventrikulären Septums.
→ Epidemiologie:
→ I: Die jährliche Inzidenz bei der hypertrophen Kardiomyopathie liegt bei 0,3-0,5/100000, wobei Männer häufiger als Frauen betroffen sind; zudem existiert eine familiäre Häufung (> 50%), sodass eine echokardiographische Screeninguntersuchung von Angehörigen empfohlen werden kann.
→ II: Sie ist die häufigste Ursache für den plötzlichen Herztod bei jungen, männlichen Sportlern; der Manifestationsgipfel der symptomatischen HCM liegt im jungen bis mittleren Erwachsenenalter (kann jedoch auch im Kindesalter auftreten).
→ Ätiologie: Die Ätiologie der hypertrophen CM ist bis heute noch nicht genau geklärt.
→ I: Die HCM wird autosomal-dominant vererbt, wobei man bei > 50% eine familiäre Häufung findet. Hierbei handelt es sich vorwiegend um Mutationen der Proteine des Sakromers. Die 3 häufigsten Mutationen findet man in folgenden Genprodukten:
→ 1) Beta-Myosin-Schwerkette (30-35% der Fälle) >
→ 2) Myosin-bindendes-Protein C (20-30%) >
→ 3) Dünnen Filamente Troponin T und I (10-15% der Fälle).
→ II: Bei der sporadischen HCM handelt es sich zum Großteil um Spontanmutationen.
→ Pathogenese:
→ I: Die Entwicklung der HCM erfolgt schon in der Fetalzeit und weist eine unterschiedliche Progression auf, weshalb sie gewöhnlich im Kindes- bis jungen Erwachsenenalter diagnostiziert wird.
→ II: Morphologisch: Es zeigt sich eine schwere, oftmals linksventrikuläre Myokard- und Septumhypertrophie (eine biventrikuläre Beteiligung ist möglich eine alleinige rechtsventrikuläre Erkrankung sehr selten), die eine Myokardversteifung und verminderte Ventrikelfüllung zur Folge hat.
→ III: Histologisch: Sind:
→ 1) Lokale Vernarbungen des Herzmuskelgewebes,
→ 2) Einengungen der kleinen, intramuralen Koronargefäße sowie
→ 3) Desorganisation der Muskelzellanordnung nachweisbar.
→ IV: Hämodynamisch:
→ 1) Gerade bei der hypertrophen-obstruktiven-CM manifestiert sich eine endsystolische Einengung der linksventrikulären Ausflussbahn mit Steigerung des intraventrikulären Druckgradienten und Ausbildung einer Mitralklappeninsuffizienz.
→ 2) Des Weiteren besteht eine diastolische Funktionsstörung mit verminderter Compliance des Ventrikels.
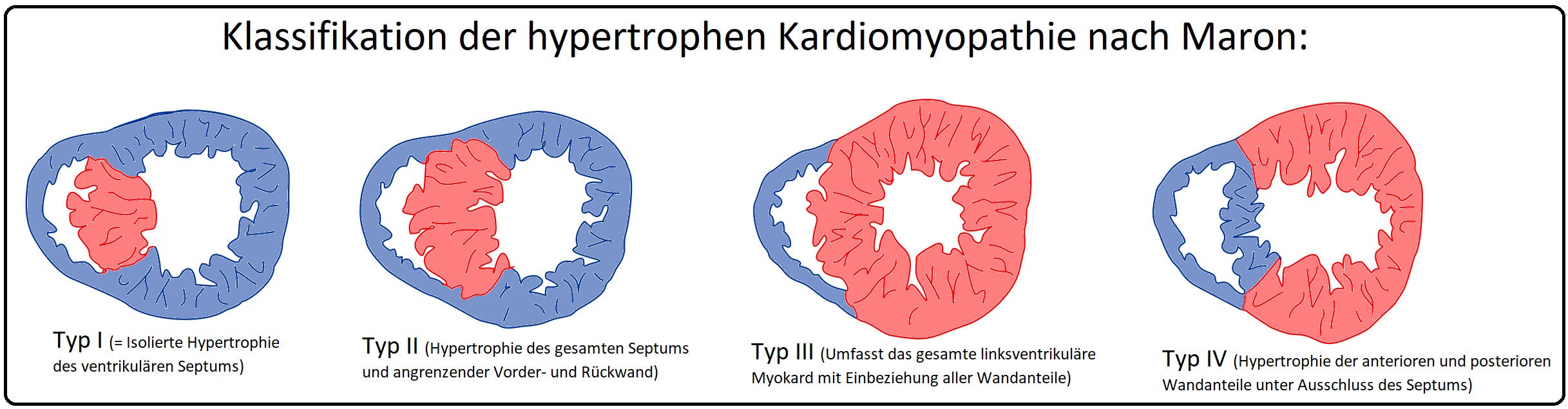
→ Klassifikation: Nach Maron unterscheidet man nach Ausprägung und Lokalisation folgende Hypertrophie-Typen:
→ I: Typ 1: Isolierte Hypertrohie des Ventrikelseptums.
→ II: Typ 2: Das gesamte Septum sowie angrenzende Teile der Vorder- und Hinterwand sind hypertrophiert.
→ III: Typ 3: Das gesamte linksventrikuläre Myokard ist hypertrophiert.
→ IV:Typ 4: Hypertrophie der anterioren und posterioren Wandanteile unter Ausschluss des Septums.
→ Klinisch-relevant: Davon abzugrenzen ist die homogene Hypertrophie des Sportlerherzens. Im Laufe des Trainings kann es über die Jahre zur Gewichtszunahme des Herzens mit Zunahme des endsystolischen und enddiastolischen Volumens kommen.
→ Klinik: Häufig ist die hypertrophe Kardiomyopathie asymptomatisch oder weist nur geringe Symptome auf; Evtl. kann der plötzliche Herztod die Erstmanifestation dieses Krankheitsbildes sein. Charakteristische Symptome der hypertrophen Kardiomyopathie sind:
→ I: Leistungsminderung und Ermüdungserscheinungen als Zeichen einer Herzinsuffizienz.
→ II: Belastungsabhängiges Dyspnoe, Palpitationen, Vorhofflimmern, insbesondere bei bradykarden oder tachykarden Rhythmusstörungen, und nicht zuletzt Angina pectoris (inkonstante Beziehung zur körperlichen Belastung oder Manifestation nach Beendigung der Belastung).
→ III: Höhergradige ventrikuläre Arrhythmien bis hin zu ventrikulären Tachykardien (und Kammerflattern, Kammerflimmer) mit Schwindel, Synkopen und plötzlichem Herztod.
→ IV: Weitere Komplikationen: Sind insbesondere:
→ 1) Infektiöse Endokarditis,
→ 2) Systemische Embolien (z.B. Lungenembolie).
→ Klinisch-relevant: Besonders bei jüngeren Menschen sollte bei Dyspnoe und Angina-pectoris-Attacken immer an eine hypertrophe Kardiomyopathie gedacht werden.
→ Diagnose:
→ I: Anamnese: Mit Familienanamnese; Eruierung eines plötzlichen Herztodes in der Familie sollte an eine hyperthrophe CM denken lassen.
→ II: Klinische Untersuchung:
→ 1) Palpatorisch ist ein breiter, hebender, nach lateral verlagerter Herzspitzenstoß nachweisbar.
→ 2) Auskultatorisch lässt sich insbesondere bei der hypertrophen-obstruktiven CM ein hochfrequentes Systolikum (Abb.: Differenzialdiagnosen Systolikum) mit Punktum maximum über dem Erb-Punkt (= 3. ICR parasternal links) nachweisen, welches durch Anstrengung oder das Valsalva Manöver verstäkrt wird; Insbesondere bei hohem Druckgradienten kann der 2. Herzon paradox gespalten sein; evtl. ist auch ein 3. und 4. Herzton eruierbar. Bei der hypertrophen-nicht-obstruktiven CM ist charakteristischerweise kein Herzgeräusch zu eruieren.
→ III: EKG: (geringe Sensitivität und Spezifität) Siehe EKG-Befund: Hypertrophe Kardiomyopathie.
→ IV: Echokardiographie: Ist bei der Diagnose-Stellung das Mittel der Wahl zur Ermittlung der Hypertrophieverteilung, Quantifizierung der Obstruktion, der systolischen und diastolischen Ventrikelfunktion sowie möglicher Begleitbefunde:
→ 1) Asymmetrische Verdickung des Ventrikelseptums, evtl. Verdickung des gesamten Ventrikelmyokards.
→ 2) Septumdicke > 13mm,
→ 3) Sanduhrförmige Einengung der linksventrikulären Einflussbahn.
→ 4) Nachweis einer Mitralklappeninsuffizienz durch Vorwölbung des vorderen Segelanteils während der Systole (SAM= systolic-anterior-movement).
→ V: MRT: Zur Bestimmung der Anatomie und Funktion des Herzens, sowie zur Druckgradienten-Bestimmung.
→ VI: Linksherzkatheter: Wenn die Diagnose mittels Echo nicht exakt gestellt werden kann, evtl. zur Endomyokardbiopsie zum Ausschluss einer Speicherkrankheit.
→ VII: Myokardbiopsie: Nachweis einer Hypertrophie bzw. eines Strukturverlustes der Myozyten und Myofibrillen, sowie eine interstitielle Fibrose. Gerade bei der HCM ohne Obstruktion wird diese häufiger durchgeführt, da in bis zu 5% eine gleichzeitige Speicherkrankheit besteht.
→ Differenzialdiagnose: Von der hypertrophen Kardiomyopathie müssen insbesondere nachfolgende Erkrankungen abgegrenzt werden:
→ I: Sekundäre Hypertrophie bei arterieller Hypertonie und Aortenstenose.
→ II: Subvalvuläre Aortenstenose,
→ III: Speicherkrankheiten wie Amyloidose, Glykogenosen oder ein kardialer Morbus Fabry.
→ Therapie:
→ I: Allgemein: Vermeiden von schweren körperlichen Belastungen aufgrund der erhöhten Gefahr eines plötzlichen Herztodes.
→ II: Medikamentös:
→ 1) Bei bestehendem Vorhofflimmern ist eine Antikoagulationstherapie (z.B. Marcumar) indiziert.
→ 2) Gabe eines Kalziumantagonisten vom Verapamil-Typ (z.B. Verapamil 240-480mg/d) oder eines Beta-Blockers.
→ 3) Besteht eine Lungenstauung in Folge eines Rückwärtsversagens des linken Ventrikels, kann mit einem Diuretika (z.B. Torasemid 5-20mg/d) begonnen werden.
→ Klinisch-relevant:
→ A) Die gleichzeitige Gabe eines Beta-Blockers und Kalziumantagonisten ist wegen eines höhergradigen AV-Blocks kontraindiziert.
→ B) Bei der hypertrophen-obstruktiven CM ist die Gabe von positiv ionotropen Substanzen (wie Digitalis, Sympathomimetikum), sowie starke Nachlastsenker, Nitrate, ACE-Hemmer aufgrund der Verstärkung der systolischen Stenose kontraindiziert.
→ III: Implantation: Eines ICD´s; insbesondere bei malignen Herzrhythmusstörungen mit ausgeprägter Linksherzhypertrophie, Synkopen und plötzlichem Herztod in der Familie. Wichtige Indikationen sind u.a.:
→ 1) Linksventrikuläre Septumdicke > 30mm,
→ 2) Fehlender/unzureichender RR-Anstieg während der Ergometrie,
→ 3) Plötzliches Herztodfälle in der Familie,
→ 4) Ventrikuläre Tachykardien im EKG,
→ 5) Rezidivierende Synkopen.
→ IV: Interventionelle Therapie:
→ 1) Perkutane transluminale-septale-Myokardablation: Transluminaler Verschluss eines Septalastes der linken Koronararterie durch exakte Alkoholinjektion. Hierdurch wird eine gezielte Myokardnekrose ausgelöst. Die Erfolgschancen hierbei liegen bei > 90%. Eine schwerwiegende Komplikation hierbei ist das Auslösen eines trifaszikulären atrioventrikulären Blocks (EKG-Befund: AV-Block).
→ 2) Transaortale subvalvuläre Myektomie: Heutzutage Standardverfahren bei Versagen der anderen Therapieverfahren.
→ V: Ultima ratio ist die Herztransplantation.
→ Prognose: Ohne Therapie liegt die jährliche Sterberate bei 1%, bei symptomatischen Patienten sogar bei 2,5% der Fälle. Besonders gefährdet an einem plötzlichen Herztod zu sterben, sind junge Männer.