→ Definition: Bei epileptischen Anfällen handelt es sich um kurzdauernde plötzlich auftretende (abnorme) exzessive Entladungen von kortikalen Neuronengruppen, die synchronisiert ablaufen. Je nach Lokalisation und Größe des Areals variieren die klinischen Phänomene, sind nicht ausschließlich motorischer Natur, gehen nicht immer mit einer Bewusstseinseintrübung einher und sind überwiegend selbstlimitierend. Die Epilepsie ist wiederum als rezidiverende epileptische Anfälle definiert. Ursachen sind insbesondere Funktionsstörungen bzw. anatomische Veränderungen es Gehirns.

→ Epidemiologie: Epilepsien stellen in Deutschland eine häufige neurologische Erkrankung (1% der Bevölkerung) dar.
→ I: Bei der Epilepsie existieren 2 Manifestationsgipfel:
→ 1) Bei Kindern und Jugendlichen mit abnehmender Prävalenz mit zunehmendem Alter und
→ 2) Bei Erwachsenen mit ansteigender Prävalenz mit zunehmendem Alter.
→ II: Erblichkeit: Das Epilepsierisiko für Nachkommen eines Elternteils für eine genetische Form liegt bei 4%, bei beiden Elternteilen mit Epilepsie steigt es auf 25%.
→ III: Das Risiko, an einem einmaligen epileptischen Anfall im Leben zu leiden, liegt deutlich über 10%, wobei der Grand-mal-Anfall die häufigste Epilepsieform darstellt.
→ Ätiologie: Epilepsien sind Erkrankungen der Großhirnrinde und können durch subkortikale Strukturen moduliert werden (jedoch laufen sie nicht in subkortikalen Strukturen, Hirnstamm oder Kleinhirn ab). Hierbei werden bezüglich der Ätiologie verschiedene Formen unterschieden:
→ I: Idiopathische Epilepsien: Sie sind genetisch bedingte Anfälle, überwiegend altersbedingt (Säuglings- und Kindesalter), werden zumeist polygen (seltener monogen vererbt und machen 40-50% der Fälle aus). Es kommt durch autosomsal-dominante Vererbung zu Defekten der Ionenkanäle (insbesondere Na+, K+- Cl-- und Ca2+-Kanäle) oder zu einem Funktionsverlust des GABA-A-Rezeptors. Hierbei existiert auch eine familiäre Häufung.
→ II: Symptomatische Epilepsie: (= Strukturell-metabolische Epilepsie) Bei dieser Form existieren exogen nachweisbare Faktoren durch akute oder chronische Hirnschädigung. Hierzu zählen insbesondere:
→ 1) Perinatale Schädigung wie perinatale Hirnschäden und Asphyxie.
→ 2) Raumfordernde Prozesse: Wie v.a. niedrig maligne (Grad I/II) Tumoren, Hydrozephalus sowie Fehlbildungen, aber auch Schädel-Hirn-Trauma (posttraumatisch mit glialer Narbenbildung), intrakranielle Blutungen, Subarachnoidalblutungen, subdurales Hämatom, Sinusvenenthrombose, karvernöse Hämangiome, Phakomatosen.
→ 3) Weitere zerebrale Veränderungen wie Durchblutungsstörungen, Apoplexie (Post-stroke-Epilepsie bei bis zu 5%; hierbei korreliert die Schwere des neurologischen Defizites positiv mit dem Risiko einer sich entwickelnden Epilepsie), hirnatrophische Prozesse, Leukodystrophie.
→ 4) Infektionen: Wie Herpes-Enzephalitis oder Creutzfeldt-Jakob-Krankheit.
→ 5) Intoxikationen: Wie Alkoholismus, Delirium tremens, Alkoholentzug, Drogenabhängigkeit und -entzug.
→ 6) Systemerkrankungen und metabolische Störungen wie Hypoglykämie, Hyperglykämie, Urämie, Addison-Krise, Phenylketonurie, Porphyrie, Schwartz-Bartter-Syndrom, Pyridoxinmangel, etc.
→ 7) Weitere Ursachen: Vaskulitis, kardiovaskuläre Störungen (Schock, hypertensive Enzephalopathie), Eklampsie, perioperativ oder nach Organtransplantationen (v.a. Herz und Leber).
→ III: Kryptogene Epilepsie: Bei dieser Form sind keine organischen oder metabolischen Ursachen zu eruieren (= ohne fassbare Ursache).

→ Pathophysiologie: Neurophysiologisch kommt es bei den epileptischen Anfällen zu einer Veränderung des Membranpotenzials mit spontaner Depolarisation und konsekutiver Entwicklung von Entladungsserien. Pathophysiologische Ursachen sind insbesondere eine Abnahme der GABA-ergen Hemmvorgänge zwischen den Nervenzellen oder eine Zunahme der exzitatorischen Transmittersubstanz Glutamat. In diesem Zusammenhang breitet sich eine spontane Depolarisation durch abgeschwächte bzw. fehlende Inhibition in der Nachbarschaft, später auch über weitere Distanz aus. Sind genügend neuronale Zellen in die synchrone Depolarisation einbezogen ist dies im EEG eruierbar.

→ Klinisch-relevant: Summarisch manifestiert sich eine extreme abnorme Synchronisierung der Neuronenaktivität im Cortex (Gruppen von Neuronen arbeiten physiologischerweise asynchron).
→ Klinik: Der epileptische Anfall ist sehr vielfältig in seiner Symptomatik (kurz, < 2min).
→ I: Hauptsymptom der Epilepsie ist der epileptische Anfall, der wiederum ein Epiphänomen eines erkrankten Hirnareals ist. So zeigt eine Erkrankung des mesialen Temporallappens (limbisches System, Hippocampus) neben den epileptischen Anfällen auch kognitive, psychische und reproduktive Störungen.
→ II: Kernsymptome: Der epileptischen Anfälle umfassen:
→ 1) Initial vor der iktalen Symptomatik (vor dem eigentlichen Anfall) sogenannte Aura als passagere Erscheinung.
→ 2) Eigentliche iktale Symptomatologie sowie
→ 3) Postiktale Erscheinungen, die sich dem Anfall anschließen.

→ III: Weitere Symptome: Jenseits der Anfälle insbesondere bei der mesialen Temporallappenepilepsie (limbisches System, Hippocampus und Amygdala) zeigen sich Symptome wie:
→ 1) Kognitive Defizite mit Abnahme der Gedächtnisfunktion für verbale und räumliche Inhalte.
→ 2) Sexuell-reproduktive Störungen mit Impotenz bei Männern und Libidoverlust sowie Anovulation (polyzystisches Ovarialsyndrom durch Störung der hypothalamischen-hypophysären-Achse) bei Frauen.
→ 3) Psychische Störungen: Bei denen v.a. die Depression im Vordergrund steht.
→ IV: Schließlich unterscheidet man bei den epileptischen Anfällen klinisch noch zwischen
→ 1) Fokalen Anfällen: Sie sind auf eine Hirnregion der Cortex beschränkt und die Symptomatik entspricht der Hirnfunktion des betroffenen Areals (mit/ohne Bewusstseinsstörung) und
→ 2) Generalisierte Anfällen: Hierbei sind alle Hirnregionen der Cortex betroffen und es besteht immer eine Bewusstseinsstörung.

→ Komplikationen: Wichtige und z.T schwerwiegende Komplikationen bei der Epilepsie sind insbesondere:
→ I: Status epilepticus: Als persistierender Anfall > 5min (Grand-mal) oder dichtes Aufeinanderfolgen von Anfällen, ohne dass der Patient das Bewusstsein wiedererlangt. Dies stellt einen lebensbedrohlichen Zustand dar (insbesondere bei Grand-mal-Status).
→ II: Eine mögliche iktogene Hypoxie kann zu morphologischen Hirnveränderungen wie Hirnatrophie, lobuläre Kleinhirnatrophie, etc. führen.
→ III: Epileptische Wesensveränderungen: Vielen oder schweren Anfällen können psychische Veränderungen wie psychomotorische Verlangsamung, Umständlichkeit im Denken und Handeln und Demenz hervorrufen; es sind sogar postiktale Psychosen möglich sowie erhöhte Suizidalität. Weitere psychische Komorbiditäten sind Angststörungen sowie Depression.
→ IV SUDEP: (= sudden unexpected death in epilepsy) 3-20-fach erhöhtes Risiko für plötzlichen Tod durch z.B. Herz- oder Atemstillstand. Das größte Risiko besteht bei Grand-mal oder medikamentenrefraktärer Epilepsie und je jünger der Patient ist umso größer das Risiko.
→ V: Weitere Komplikationen: Sind u.a.:
→ 1) Sekundäre Traumata durch den Krampfanfall selbst infolge z.B. eines Sturzes (Wirbelkörperfraktur).
→ 2) Des Weiteren durch z.B. Aspiration, Hypoxie, etc.
→ Diagnose: Grundsätzlich setzt die Diagnose der Epilepsie das Vorhandensein eines epileptischen Anfalls voraus. Wichtige hierbei ist die umfangreiche Anamnese:
→ I: Anamnese/klinische Untersuchung: Erfolgt in Eigen- und Fremdanamnese und umfasst:
→ 1) Mit Anfallsanamnese (subjektive Anfallsschilderung, Zeitpunkt der Anfallsereignisse (Schlaf, Aufwachphase), Provokationen bzw. Triggermechanismen, Vorboten (= sogenannte Aura mit z.B. epigastrische Missempfindungen, Schwindel, Deja-vu-Erlebnissen, etc.), aber auch Bewusstlosigkeit, Amnesie über den Anfall, Verletzungen, Zungenbiss, Urin- oder Kotabgang, postiktale Symptome, etc. Ergänzt wird sie durch eine mögliche Fremdanamnese von Personen, die den Anfall beobachtet haben (z.B. Dauer des Anfalls, anfallsassoziierte neurologische Symptome).
→ 2) Geburtsanamnese: Mit perinantaler Schädigung, Fieberkrämpfe, frühe Schädel-Hirn-Traumen, Meningitis, Enzephalitis und weitere neurologische Erkrankungen.
→ 3) Ausschluss internistischer Erkrankungen, Alkohol- und Drogenabhängigkeit sowie die Eruierung der Medikamenteneinnahme und nicht zuletzt
→ 4) Die familiäre Disposition.

→ II: EEG: (Enzephalographie) Das EEG ist eine wichtige Zusatzdiagnostik bei der Epilepsie. Im ersten EEG nach einem epileptischen Anfall lassen sich überwiegend keine epilepsietypischen Potenziale nachweisen. Die Sensitivität des EEG nimmt erst mit der Zahl der Untersuchungen zu (nach dem 4-6 EEG liegt sie in den Folgemonaten bei 60-80%). Auch ist die Detektion der epilesietypischen EEG-Veränderungen innerhalb der ersten 12-24 Stunden am höchsten (jedoch lassen sich auch im anfallfreien bzw. interiktalen Intervall Potenzialänderungen darstellen).Wichtige epilepsietypische Potenziale des EEG sind u.a.:
→ 1) Spikes: Schnelle und langsame Wellen.
→ 2) Spike-Waves-Paroxymen: Spitzen und Wellen.
→ 3) Sharp-Waves: Steile Wellen.
Beweisend für die Epilepsie ist insbesondere eine Video-EEG-Registrierung (einschließlich des iktalen EEG-Musters). Zudem haben sich sogenannte Provokationsmanöver zur Steigerung der Sensitivität von EEG-Ableitungen etabliert:

→ Klinisch-relevant: Mit Hilfe des EEG kann man sowohl bei der generalisierten als auch bei der fokalen Form Hinweise für das Epilepsie-Geschehen eruieren.
→ A) So sind bei den generalisierten Epilepsien die Frequenz der Spike-Wave-Komplexe sehr unterschiedlich.
→ 1) 2-2,5Hz sind charakteristisch für symptomatische generalisierte Epilepsien.
→ 2) 3Hz sind typisch für Absence-Epilepsie und
→ 3) Hochfrequente Spike-wave-Komplexe über 3,5 Hz sind wiederum charakteristisch für die juvenile myoklonische Epilepsie.
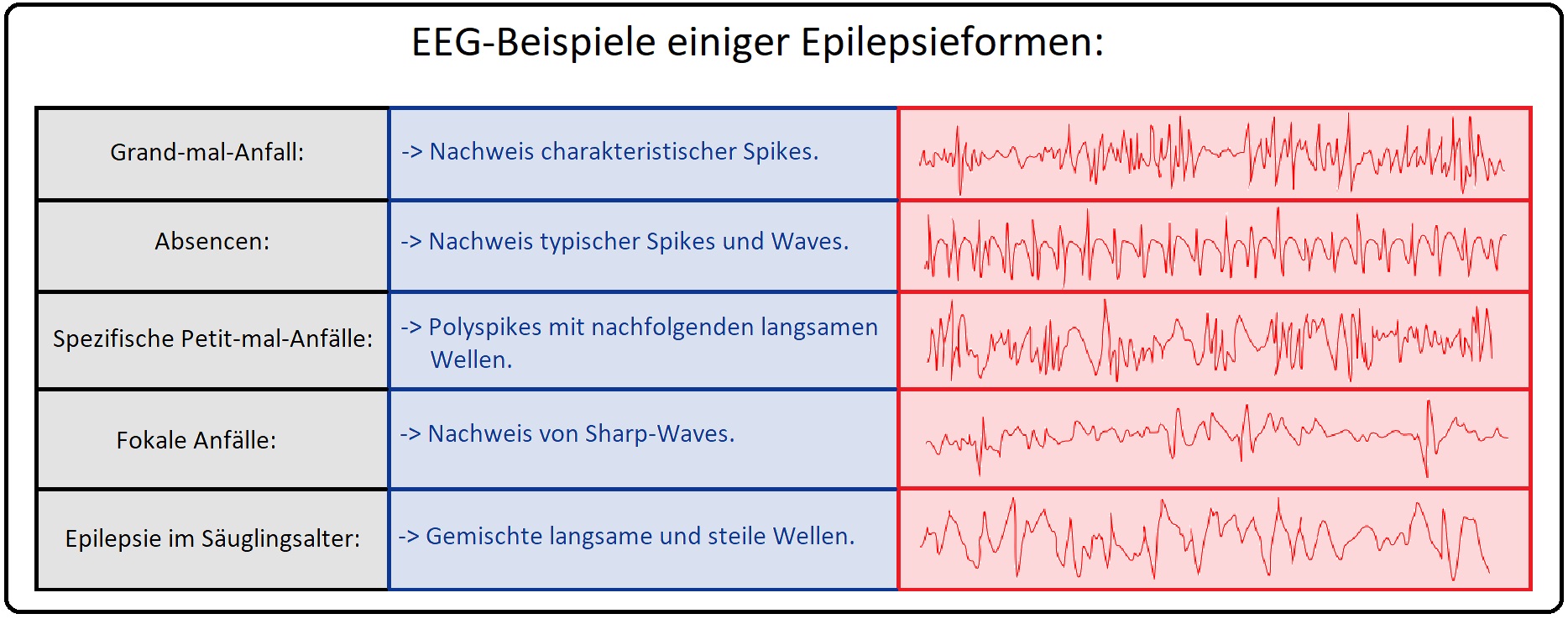
→ B) Bei den fokalen Epilepsien gibt die Lokalisation der fokal interiktalen Potenziale Auskunft über das Epilepsiesyndrom.
→ C) Epilepsietypische Potenziale sind nicht beweisend für das Vorliegen einer Epilepsie. So können neurologische Erkrankungen wie z.B Schlaganfall, intrazerebrale Blutungen, Morbus Alzheimer oder Hirntumoren zum Auftreten von epilepsietypischen Potenzialen führen, ohne dass eine Epilepsie vorliegt (= epilepsietypische Potenziale stellen keinen Beweis für eine Epilepsie dar).
→ III: Bildgebung:
→ 1) MRT: Die MRT stellt ist Goldstandard bei der Diagnose der Epilepsie und dient der Darstellung struktureller Gehirnveränderung. Es gibt jedoch MRT-Befunde, die häufig bei symptomatischen Epilepsien nachzuweisen sind; hierzu zählen insbesondere Ammonshornsklerose (= Hippocampussklerose), Kavernome (häufig mit Blutungszeichen), Hamartome, tuberöse Sklerose, etc.
→ 2) CT: Ein cCT ist nur indiziert, wenn in der postiktalen Phase neurologische Defizite bestehen und eine Akutsituation wie eine intrazerebrale Blutung ausgeschlossen werden muss.
→ 3) Nuklearmedizinische Verfahren: Wie PET und SPECT die ausschließlich der prächirurgischen Diagnostik vorbehalten sind. Sie stellen graphisch die Gehirnfunktionen dar bezogen auf den Blutfluss (SPECT) und den Metabolismus (PET). Epileptische Areale zeigen im interiktalen Intervall eine Hypoperfusion und im Anfall eine Hyperperfusion sowie einen Hypo- bzw. Hypermetabolismus im PET.
→ IV: Labor: Hierbei steht insbesondere die Bestimmung der Keratinkinase (CK) mit Werte bis über 1000U/l innerhalb der ersten 24 Stunden bei generalisierten tonisch-klonischen Anfällen im Vordergrund. Die Bestimmung des Prolaktins mit transientem Anstieg (0-30min postiktal) wird insbesondere bei der Temporallappenepilepsie empfohlen. Die Liquoruntersuchung ist wiederum bei Verdacht auf ein infektiöses Geschehen (z.B. Enzephalitis) indiziert.

→ Differenzialdiagnose: Von der Epilepsie müssen insbesondere nachfolgende Erkrankungen abgegrenzt werden:
→ I: Drop-Attack mit plötzlichem Sturz, jedoch ohne Bewusstseinsverlust.
→ II: Paroxysmale Dyskinesien weisen charakteristischerweise dystone oder choreoathetotische Abläufe ohne Bewusstseinsverlust und nur von kurzer Dauer auf (z.T. schwierig von Frontallappenanfällen zu unterscheiden).
→ III: Narkolepsie: Die betroffenen Patienten sind jederzeit erweckbar.
→ IV: Psychogene Anfälle mit polymorpher Klinik wie Areaktivität, hypermotorischer Bewegung und einer statusartigen Dauer (> 5min). Hierbei existieren jedoch situative Triggermechanismen und das EEG ist unauffällig.
→ V: Der Hyperventilationstetanie geht zumeist ein Gefühl der Atemnot voraus und es besteht kein Bewusstseinsverlust (die Hyperventialtionstetanie kann jedoch auch einen epileptischen Anfall triggern).
→ VI: Synkopen (z.B. vasovagale, kardial bedingte, etc.) weisen eine typische Triggersituation mit „Schwarz-werden vor den Augen“ auf. Der Bewusstseinsverlust ist von kurzer Dauer und es besteht keine Verlangsamung und Verwirrtheit.
→ VII: Weitere Differenzialdiagnosen: Sind insbesondere:
→ 1) Eine Aura kann sich auch im Rahmen einer Migräne manifestieren.
→ 2) Einschlafmyoklonien oder REM-Schlaf-Verhaltensstörungen (= Parasomnie mit Unruhe, komplexen Bewegungen zumeist in der 2. Nachthälfte).
→ 3) Hirnstammanfälle bei multipler Sklerose mit plötzlichen Muskelkontraktionen einer Körperhälfte bei erhaltenem Bewusstsein und ohne EEG-Veränderungen.
→ 4) Des Weiteren transitorische globale Amnesie, Katalepsie, Panikattacken, etc.
→ 5) Bei Kleinkindern Fieber- und Zahnkrämpfe.

→ Therapie: Die Behandlung der Epilepsie ist selten eine kausale oder kurative, sondern vielmehr eine chronisch antikonvulsive Prophylaxe und umfasst:
→ I: Allgemeine Maßnahmen: Insbesondere die Vermeidung anfallsauslösender Situationen wie Extrembelastungen, Schlafmangel, Alkohol, Flackerlicht, Therapie mit Kortikosteroiden (z.B. bei kindlichem West-Syndrom oder einer Epilepsie aufgrund einer limbischen Enzephalitis), etc.
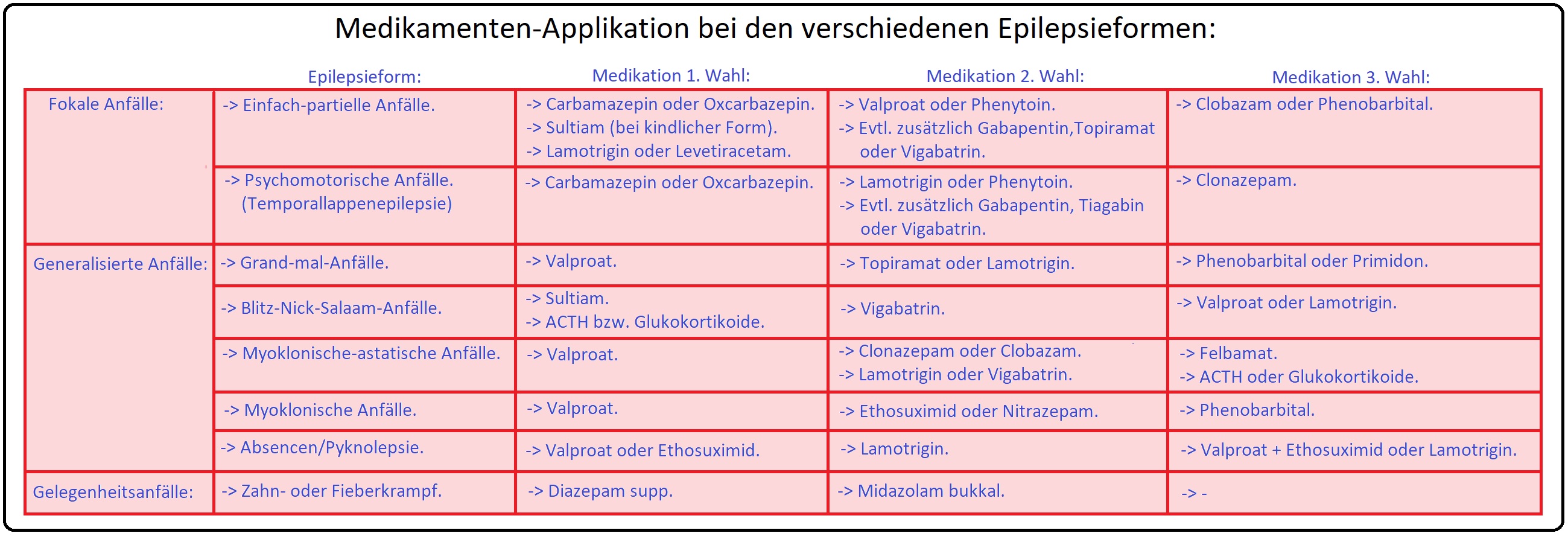
→ II: Medikamentöse Therapie: Eine Indikation für die medikamentöse Therapie der Epilepsie ist das manifeste Anfallsleiden, d.h. mindestens 2 Anfälle innerhalb von 6 Monaten oder ein einmaliger Anfall mit pathologischem EEG. Hierbei handelt es sich um eine antikonvulsive Prophylaxe zur Reduktion bzw. Verhinderung epileptischer Anfälle. Die Entscheidung für eine antiepileptische Therapie hängt insbesondere von der Schwere des ersten Anfalls bzw. Anfälle, anfallsbedingten Verletzungen, der Angst vor Rezidiven sowie der sozialen Konsequenzen (Fahr- und Berufseignung) und dem Letalitätsrisiko ab. Jedoch sind bei der medikamentösen Therapie der Epilepsie nachfolgende Grundprinzipien zu beachten:
→ 1) Aufdosierung einer ausgewählten Substanz bis zur Anfallsfreiheit oder bis zum Auftreten von intolerablen Nebenwirkungen (im Vordergrund steht die Klinik weniger der Serumspiegel).
→ 2) Bei fehlender Anfallsfreiheit unter der gewählten Medikation ist eine überlappende Umstellung auf eine weitere Monotherapie (Ausnahme Umstellung von Carbamazepin auf Oxcarbazepin) indiziert.
→ 3) Nur bei fehlender Anfallsfreiheit erfolgt eine Kombinationstherapie aus zwei Antikonvulsiva in der Regel mit unterschiedlichem Wirkungsmechanismus (auf unterschiedliche Ionenkanäle der Nervenzellen).
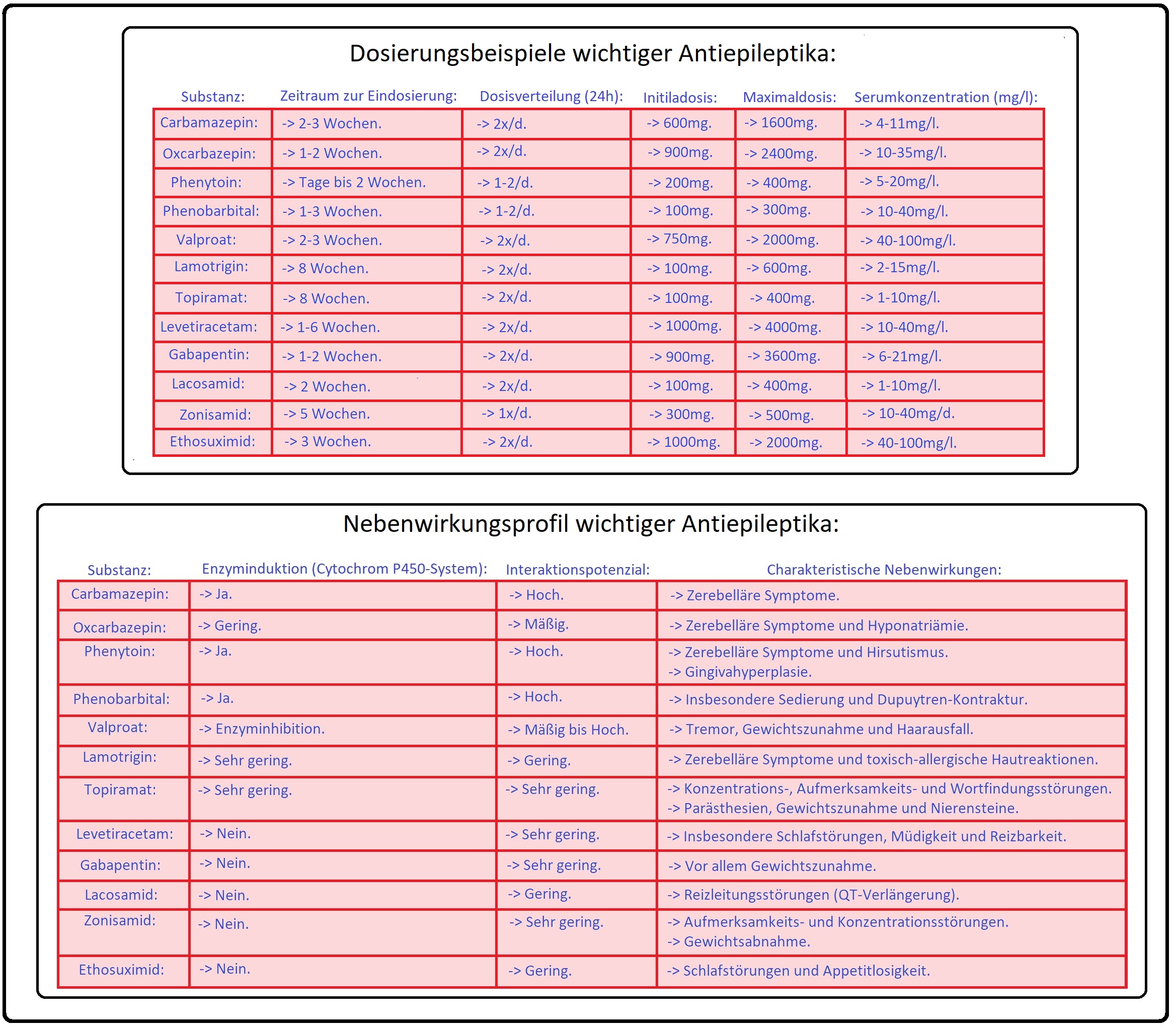
→ II: Medikamentenauswahl: Die Auswahl der Antikonvulsiva wird immer individuell getroffen und folgt primär den pathophysiologischen Gegebenheiten. So haben sich bei den generalisierten Anfällen insbesondere Antikonvulsiva wie Valproat (Mittel der Wahl zur Therapie der generalisierten Anfälle vom idiopathischen Typ), Lamotrigin (zu Monotherapie von symptomatischen Anfällen), Levetiracetam (weist insbesondere in der Kombinationstherapie von symptomatischen Anfällen die höchste Wirksamkeit), Topiramat, etc. durchgesetzt. Die weitere Auswahl erfolgt anschließend nach Kriterien wie Vermeiden von Nebenwirkungen, Interaktionen, Compliance, Kosten, etc. Des Weiteren können individuelle Faktoren die Medikamentenauswahl zusätzlich beeinflussen; hier zählen u.a.:
→ 1) Rasches Erreichen der Zieldosis bei Topiramat und Levetiracetam.
→ 2) Geringe Interaktion mit weiteren Medikamenten bei Lamotrigin, Levetiracetam oder Topiramat.
→ 3) Geringe kognitive Nebenwirkungen bei Valproat und Lamotrigin.
→ 4) Gewichtsneutralität insbesondere bei Carbamazepin, Lamotrigin und Oxcarbazepin.
→ 5) Keine reduzierte Wirkung von hormonellen Kontrazeptiva durch z.B. Valproat oder Levetiracetam, denn enzyminduzierende Antikonvulsiva wie z.B. Carbamazepin, Oxcarbazepin, Primidon, Topiramat (Dosis > 200mg/d) etc. setzten die empfängnisverhütende Wirkung deutlich herunter.
→ 6) Geringe Teratogenität bei Lamotrigin und Carbamazepin.
→ Klinisch-relevant: Wichtige Wirkungsmechanismen der Aniepileptika sind u.a.:
→ A) Membranstabilisierend wie Carbamazepin und Phenytoin.
→ B) Erhöhung der GABA-Konzentration mit konsekutiver Hemmung der Neuronenaktivität durch z.B. Benzodiazepine, Barbiturate, Valproat oder Tiagabin.
→ C) Glutamatantagonisten wie Gabapentin, Lamotrigin, Topiramat, etc.
→ III: Zieldosis/Kombinationstherapie: Allgemein kann festgehalten werden, das die Latenz bis zum vollständigen Wirkungseintritt und konsekutiven Steady-state bis zu 2 Monaten dauern kann, sodass ein schneller Wechsel aufgrund der Wirkungslatenz nicht empfohlen wird. In diesem Zusammenhang beurteilt man den antikonvulsiven Effekt der Substanz nach dem vierfachen zeitlichen Intervall (1 Anfall pro Woche = Beobachtungszeit 4 Wochen). Zeigt sich hierbei eine unzureichende Wirkung kann die Dosis gesteigert auf ein weiteres Medikament gewechselte oder eine zusätzliche Substanz addiert werden. Insbesondere wenn die Anfallsfreiheit mittels Monotherapie nicht gelingt, wird heutzutage zumeist ein Standardantiepileptikum (z.B. Carbamazepin oder Valproat) mit einem Antiepileptikum der neuen Generation (z.B. Gabapentin, Vigabatrin, Lamotrigin, Tiagabin, etc.) kombiniert; dies wird als sogenannte "Add-on-Medikation" bezeichnet. In diesem Zusammenhang haben sich u.a. nachfolgende Antiepileptikakombinationen etabliert:
→ 1) Antiepilepsiekombination zur Therapie symptomatischer Epilepsie mit Lamotrigin + Valproat oder Lamotrigin + Levetriacetam oder Carbamazepin + Levetriacetam.
→ 2) Generalisierte Anfälle bei idiopathischer Epilepsie mit Valproat + Lamotrigin oder Valproat mit Levetriacetam.
→ IV: Therapieende: Nach 2-5 Jahren Anfallsfreiheit und unauffälligen EEG-Kontrollen kann ein Absetzen der antiepileptischen Medikation in Erwägung gezogen werden, jedoch müssen die Substanzen (aufgrund der langen Applikation) ausgeschlichen werden. Manifestiert sich allerdings ein Rezidiv (in 1/3 der Fälle) ist eine lebenslange Pharmakotherapie indiziert.
→ V: Weitere Therapien: Hierzu zählen insbesondere:
→ 1) Epilepsiechirurgie: Die chirurgische Indikationen sind insbesondere fokale Epilepsien, die gegenüber mindestens 2 Substanzen (in maximaler Dosierung) pharmakorefraktär sind, eine hohe Anfallsfrequenz, einen eingrenzbaren Epilepsiefokus, eine deutliche Verbesserung der Lebensqualität (ohne z.B. nachfolgende neurologische Defizite), etc. aufweisen. Charakteristische epilepsiechirurgische Eingriffe sind z.B. die selektive Amygdala-Hippocampektomie, anterotemporale Resektion, Läsionektomie oder Lobektomie (so wird z.B. bei der Temporallappenepilepsie die Amygdala-Hippocampektomie oder die antero-temporale Resektion durchgeführt). Besonders gute chirurgische Erfolge zeigen sich bei kleinen, lokalen Ursachen wie Harmatomen, Kavernomen oder der Hippocampussklerose. Bis zu 60% der operierten Patienten sind postoperativ (unter Medikation) anfallsfrei.
→ 2) Stimulationsverfahren: Diese speziellen Verfahren werden als „palliative Säule“ ausschließlich bei therapierefraktärer Epilepsie eingesetzt. Zum einen existiert die (linke) Nervus-vagus-Stimulation“; hierbei kommt es zur 30 Minuten (30Hz) Stimulation mit anschließender 5-minütiger Pause (durch ein subkutan implantiertes Stimulationsgerät). Angenommen wird eine Modulation der Anfallsschwelle durch Stimulation der vagalen Afferenzen über aszendierende Bahnen aus dem Hirnstamm. Sie kann sowohl bei den fokalen als auch bei den generalisierten Epilepsien sowie bei Patienten mit eingeschränkter kognitiver Compliance eingesetzt werden. Es zeigt sich, dass 1/3 der Patienten eine deutliche Abnahme der Anfallsfrequenz aufweisen. Ein weiteres Stimulationsverfahren in der antikonvulsiven Therapie ist die Tiefenhirnstimulation. Hierbei erfolgt die Stimulation mittels stereotaktisch implantierter Tiefenelektroden (beidseits im anterioren Thalamus oder Hippocampus).
→ Prognose: Die Kontrolle der Anfälle ist für die meisten Patienten lebenslang und die Anfallsfrequenz pendelt sich individuell ein (fokale Anfälle häufiger pro Zeiteinheit als generalisierte Anfälle). Zudem werden bis zu 70% der Betroffenen unter einer adäquaten antikonvulsiven Therapie anfallsfrei. Jedoch weisen Patienten mit Epilepsie ein erhöhtes Mortalitätsrisiko auf; Ursachen hierfür sind insbesondere Verletzungen im Anfall, Asystolie im Anfall als „sudden unexpected death in epilepsy", Status epilepticus und nicht zuletzt die erhöhte Suizidalität bei bestehender Depression.