- Details
- Geschrieben von: CF
- Kategorie: Organische psychische Störungen
- Zugriffe: 9234
→ Definition:
→ I: Die Lewy-Body-Demenz ist definiert als ein demenzielles Syndrom, welches charakteristischerweise durch das Auftreten von fluktuierenden kognitiven Defiziten, Vigilanzstörungen und optischen Halluzinationen gekennzeichnet ist.
→ II: Schon früh in der Krankengeschichte entwickeln die Patienten ein Parkinson-Syndrom.
→ III: Ein weiteres Charakteristikum ist der Nachweis von sogenannten PAS-positiven Einschlüssen (= Lewy-Körperchen: Hierbei handelt es sich um runde eosinophile Einschlüsse im Zytoplasma der Neuronen in der Kortex, dem Hirnstamm, limbischen System, Nucleus basalis Meynert etc.).
→ Epidemiologie:
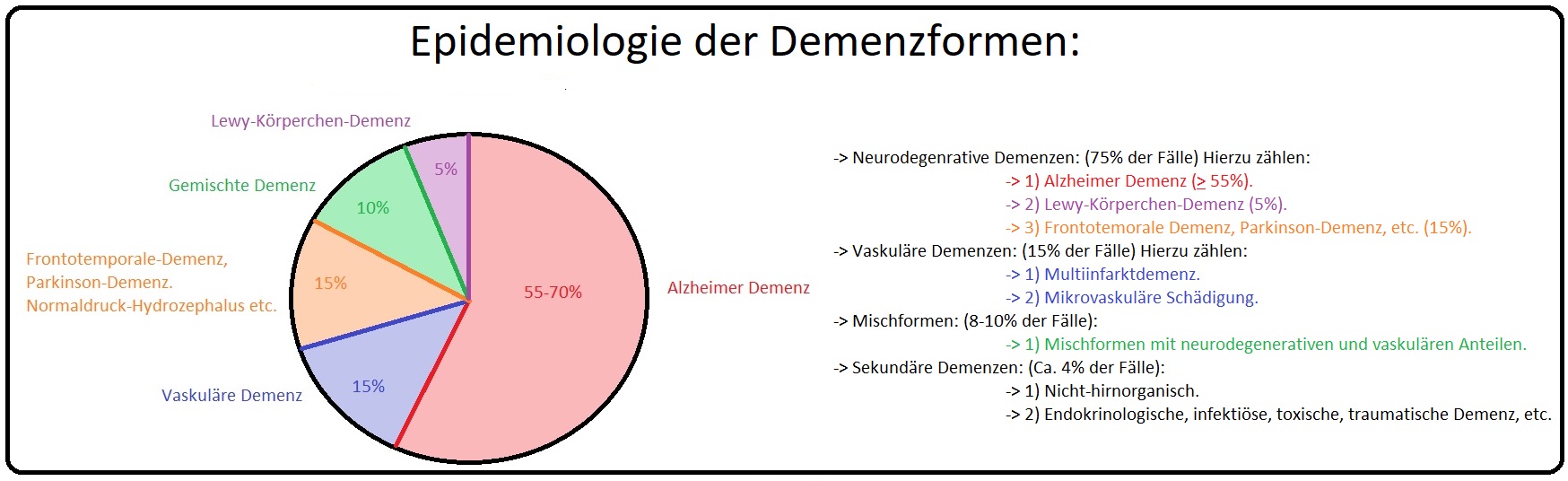
→ I: Die Lewy-Körperchen Demenz ist mit 5-15% der Fälle eine häufige Demenz-Form und hat ihren Manifestationsgipfel in der 7. Lebensdekade.
→ II: Männer sind etwas häufiger als Frauen betroffen.
→ Ätiopathogenese: Die genaue Ätiologie ist bis heute noch nicht bekannt, jedoch werden genetische Komponenten angenommen.
→ Pathologie:
→ I: Charakteristikum ist der Nachweis von Lewy-Körperchen in den kortikalen Neuronen, insbesondere der Neokortex, im limbischen System und den Basalganglien, aber auch in der Substancia nigra.
→ II: Bei den Lewy-Körperchen handelt es sich um eosinophile intraplasmatische neuronale Einschlusskörperchen, die vor allem aus aggregierten Alpha-Synuklein-Molekülen (Ursache der Aggregation ist eine Mutation im SNCA-Gen mit konsekutiver Synukeinopathie) bestehen. Diese Aggregate wirken neurotoxisch und beeinflussen insbesondere die Verteilung und den Metabolismus von Dopamin.
→ III: Weitere neuroanatomische Veränderungen: Sind u.a.:
→ 1) Senile Plaques und Neurofibrillen (= Tangles) analog zur Alzheimer-Demenz,
→ 2) Kortikale Spongiose vor allem im frontalen und temporalen Bereich.
→ Klassifikation: Je nach Lokalisation der Lewy-Körperchen im ZNS werden folgende Krankheitsbilder differenziert:
→ I: Demenz bei Morbus Parkinson: Subkortikal vor allem in der Substancia-nigra gelegene Lewy-Bodies. Ein wichtiges Unterscheidungsmerkmal zur Lewy-Body-Demenz ist, dass die extrapyramidal-motorische Symptomatik (Tremor, Rigor, Akinese) länger als 1 Jahr besteht, bevor es zum Auftreten von kongitiven Defiziten kommt.
→ II: Reine Lewy-Body-Demenz: Hier manifestieren sich die Einschlusskörperchen insbesondere im Bereich der Kortex. Bei dieser Form tritt die kognitive Symptomatik vor oder innerhalb des 1. Jahres nach Manifestation der motorischen Symptome auf. Eine Alzheimer-Pathologie fehlt.
→ III: Diffuse Lewy-Body-Demenz: Mit charakteristischen kortikaler Lokalisation und zusätzlichem Nachweis von senilen Plaques und Neurofibrillen (mit Alzheimer-Pathologie).
→ Klinik: Bei der Lewy-Body-Demenz stehen zu Beginn der Erkrankung vor allem Störungen der Aufmerksamkeit und visokonstruktive Störungen im Vordergrund.
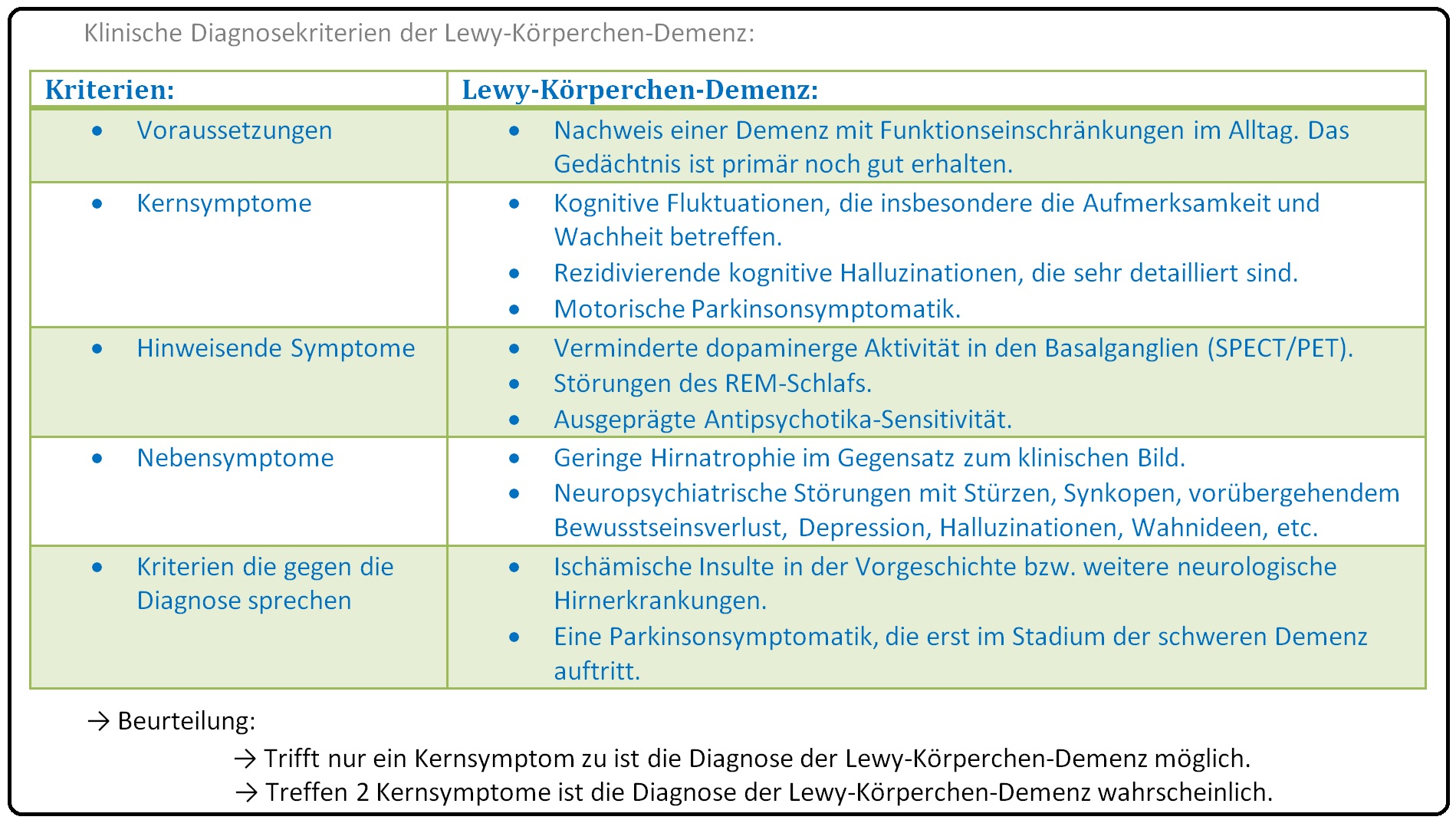
→ I: Kernsymptome:
→ 1) Stark fluktuierende kognitive Leistungsfähigkeit, insbesondere bezogen auf die Aufmerksamkeit und Wachheit mit Symptomen wie Schläfrigkeit tagsüber, Episoden vollständiger Verwirrtheit und evtl. persistierender regungsloser Starre.
→ 2) Spontane extrapyramidal-motorische Symptome mit Bradykinese und Rigor, der Ruhetremor fehlt zumeist.
→ 3) Komplex ausgestaltete, szenenhafte visuelle Halluzinationen (aber auch akustische Halluzinationen möglich), die z.T. bedrohlich erscheinen, treten meist spontan auf.
→ Klinisch-relevant: Patienten mit Lewy-Körperchen-Demenz haben eine ausgeprägte Neuroleptika-Sensitivität in Bezug auf die Entwicklung extrapyramidal-motorische Nebenwirkungen.
→ II: Nebensymptome: Hierzu zählen u.a.:
→ 1) Schenk-Syndrom: Hierbei handelt es sich um eine REM-Schlafstörung mit Sprechen und Schreien im Schlaf, sowie motorischem Ausagieren von Träumen.
→ 2) Rezidivierendes Stürzen, Bewußtseinsstörungen, Synkopen.
→ 3) Psychiatrische Symptome: Mit Depression, systematisierten Wahnideen sowie im weiteren Krankheitsverlauf auftretenden akustischen und taktilen Halluzinationen.
→ IV: Vegetative Symptome: Sind relativ häufig mit Hyperhidrosis, imperativem Harndrang/Harninkontinenz, arterieller Hypotonie, orthostatischer Dysregulation und Obstipation.
→ Diagnose:
→ I: Anamnese/klinische Untersuchung:
→ 1) Fremdanamnese mit Eruierung von Wahnideen, Halluzinationen, Nachweis kognitiver Fluktuationen.
→ 2) Klinischer Nachweis der Kernsymptome bzw. der Merkmale, die die Diagnose stützen:
→ II: Labor: Bestimmung der Glukose, Elektrolyte, Vitamin B12, Leberenzyme, Nierenretentionswerte und der Ammoniak-Konzentration gehört zu den Routinelaborparametern der Demenz.
→ III: Bildgebende Verfahren:
→ 1) cCT/cMRT: Im Vergleich zum klinischen Bild manifestiert sich nur eine geringe Hirnatrophie (beim Morbus Alzheimer deutlich ausgeprägter).
→ 2) PET/SPECT: Charakteristische Minderperfusion bzw. Hypometabolismus in den parietooczipitalen Hirnarealen, einschließlich der visuellen Primärkortex.
→ 3) Dopamin-PET: Hierbei zeigt sich eine verminderte präsynaptische Dopaminaufnahme im Striatum.
→ Differenzialdiagnose: Von der Lewy-Body-Demenz müssen insbesondere nachfolgende Erkrankungen abgegrenzt werden:
→ I: Alzheimer Demenz: Gerade im Frühstadium gestaltet sich die Unterscheidung als sehr schwierig, vor allem wenn initial die Parkinson-Symptomatik fehlt. Kognitive Fluktuationen fehlen zumeist bei der Alzheimer Demenz.

→ II: Parkinson-Demenz: Schwierig ist die Differenzierung zwischen Lewy-Body-Demenz und der Demenz bei Parkinson. Entwickelt sich die Demenz später als ein Jahr nach der Parkinson-Symptomatik spricht dies für einen Morbus Parkinson (= einziges Diagnosekriterium ist die zeitliche Abfolge von motorischen und kognitiven Störungen).
→ III: Vaskuläre Demenz: Hierbei sind die kardiovaskulären Risikofaktoren oder ein ischämischer Insult in der Vorgeschichte des Patienten eruierbar.
→ IV: Weitere Differenzialdiagnosen: Sind insbesondere
→ 1) Pseudodemenz bei schwerer Depression und
→ 2) Ganser-Syndrom, etc.

→ Therapie:
→ I: Allgemeinmaßnahmen: Umfangreiche Aufklärung und Beratung der Angehörigen.
→ II: Medikamentöse Therapie:
→ 1) Antidementiva: Wie aus Beobachtungen hervorgeht, sprechen Patienten mit Lewy-Körperchen-Demenz ausgesprochen gut auf die Behandlung mit Cholinesterase-Hemmern an. Sie stellt zur Zeit das Mittel der ersten Wahl dar; insbesondere Rivastigmin (3-6mg/d) wird v.a. zur Therapie kognitiver Defizite und Halluzinationen substitutiert (hierbei ist jedoch eine Verschlechterung der extrapyramidal-motorischen Symptome möglich).
→ 2) L-Dopa: Bei schweren extrapyramidal-motorischen Störungen ist ein Behandlungsversuch mit L-Dopa oder Amantadin zu erwägen. Sie haben jedoch ein ausgeprägtes Potenzial, Halluzinationen zu induzieren.
→ 3) Neuroleptika: Die klassischen Neuroleptika sind bei dieser Demenzform aufgrund der immensen Neuroleptika-Sensitivität kontraindiziert. Jedoch kann ein Therapieversuch mit einem atypischen Neuroleptikum z.B. Quetiapin (in einer Dosis von 12,5-100mg/d) bei schweren Halluzinationen und Wahnideen versucht werden.
- Details
- Geschrieben von: CF
- Kategorie: Organische psychische Störungen
- Zugriffe: 13198
→ Definition:
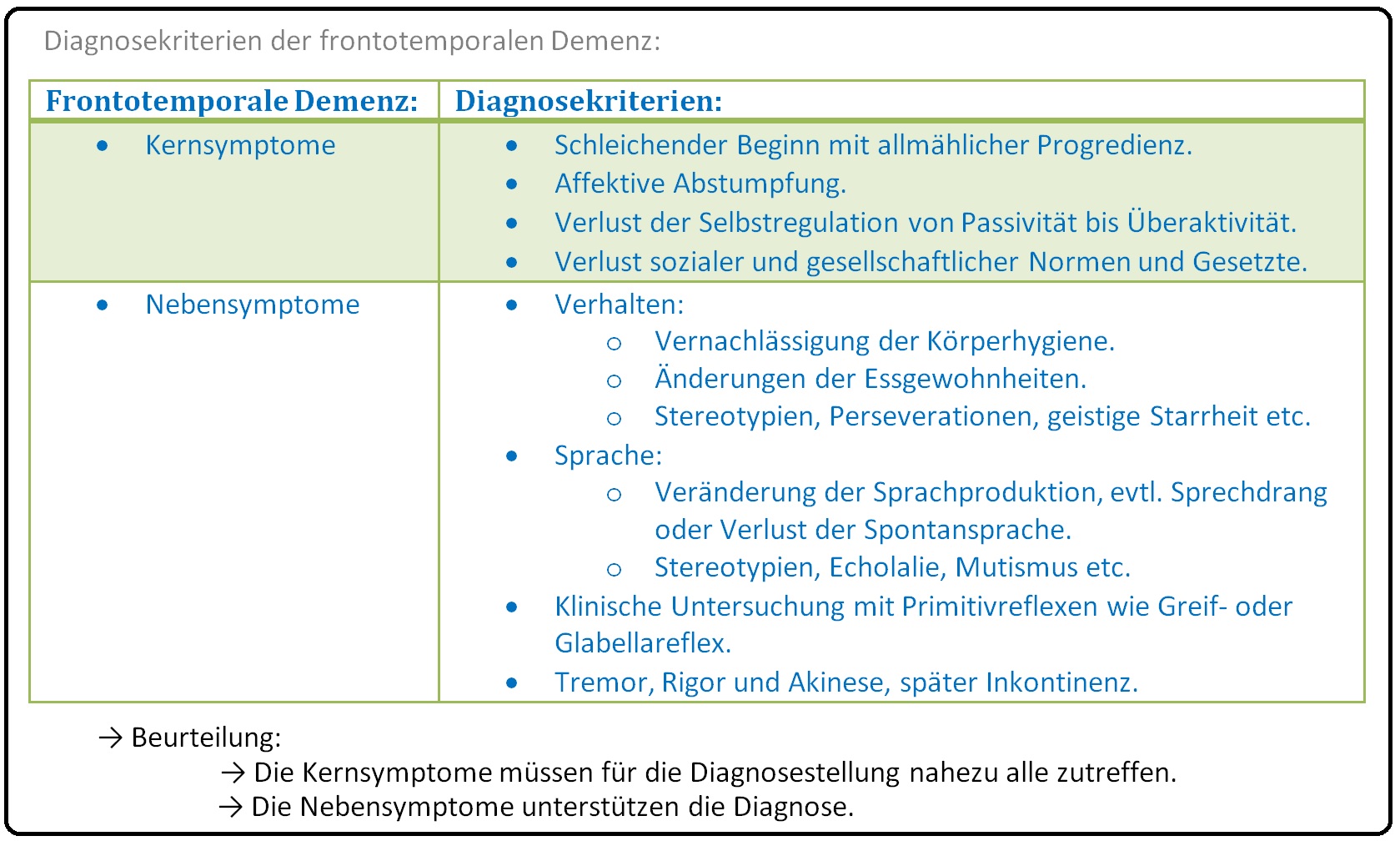
→ I: Bei der frontotemporalen Demenz handelt es sich um eine heterogene Gruppe von klinischen Syndromen, die sich durch neurodegenerative Prozesse der Neokortex, insbesondere der anterioren Anteile des Frontalhirns und des Temporallappens, charakterisieren.
→ II: Je nach Lokalisation der Degeneration werden 3 Subtypen der FTD unterschieden:
→ 1) Frontotemporale Demenz: (= früher Morbus Pick) Stellt mit 70-80% der Fälle die häufigste Form dar mit einer frontal betonten Hirnatrophie.
→ 2) Semantische Demenz: Ist in 10-20% der Fälle nachweisbar und weist eine anterior temporal betonte Hinratrophie auf sowie
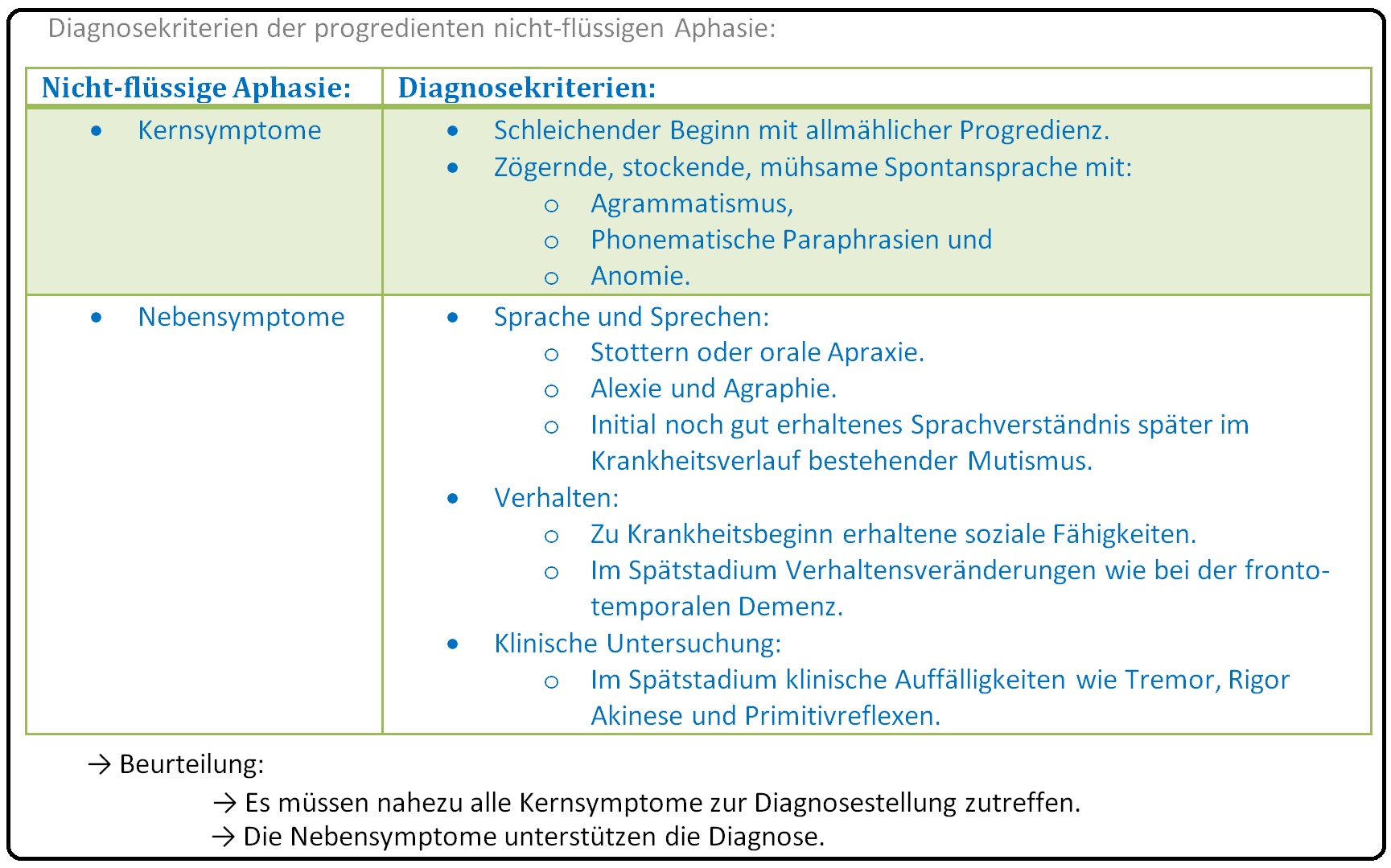
→ 3) Progrediente nicht-flüssige Aphasie: Mit 5-10% der Fälle sehr selten. Es zeigt sich eine frontolaterale Atrophie der sprachdominaten Hemisphäre.
→ Epidemiologie:
→ I: Die Inzidenz liegt bei 1-2/100000 und tritt somit deutlich seltener als die Alzheimer Demenz auf; allerdings macht die frontotemporale Demenz mehr als 20% der präsenilen Demenzen aus.
→ II: Der Manifestationsgipfel liegt zwischen dem 50.-60. Lebensjahr, wobei auch Krankheitsbeginne um das 30.-40. Lebensjahr beschrieben wurden.
→ III: Frauen sind etwas häufig als Männer betroffen.
→ Ätiopathogenese:
→ I: Zumeist entwickelt sich die frontotemporale Demenz sporadisch, in 10% der Fälle weist sie jedoch einen autosomal-dominanten Erbgang auf (Chromosom 3, 9 und 17).
→ II: Hierbei manifestieren sich gerade Mutationen auf dem Progranulin- und MAPT-Gen (kodiert das Tau-Protein) auf Chromosom 17.
→ III: Histopathologisch kristallisiert sich eine frontotemporale Hirnatrophie mit massivem Nervenzellverlust sowie kortikal geschwollene Neurone (= es handelt sich um argentophile Pick-Körpern in den Gliazellen = Pick-Zellen) heraus.
→ IV: Mittels Immunhistochemie können Tau-Proteine oder Ubiquitin-Einschlüsse nachgewiesen werden.
→ Klinisch-relevant: Alle Varianten der frontotemporalen Demenz weisen Gemeinsamkeiten auf. Hierzu zählen:
→ A) Eine frühe progressive Wesens- bzw Persönlichkeitsveränderung,
→ B) Zum Teil ausgeprägte Verhaltensauffälligkeiten (insbesondere des Sozialverhalten) und
→ C) Eine progrediente Beeinträchtigung des Sprachvermögens (und planendem Denkens).
→ Klinik: Die klinische Symptomatik richtet sich vor allem nach den pathologisch anatomischen Veränderungen. Zumeist weist sie jedoch einen schleichenden Beginn und eine langsame Progredienz auf.
→ I: Frontotemporale Demenz: Hier zeigt sich eine Degeneration der Präfrontalkortex sowie der frontalen Pole beider Temporallappen. Zu Krankheitsbeginn stehen Veränderungen der Persönlichkeit und des Verhaltens im Vordergrund. Typische Symptome sind:
→ 1) Witzelsucht,
→ 2) Verlust der Manier und sozialer Fähigkeiten,
→ 3) Triebhafte Enthemmung und Distanzlosigkeit,
→ 4) Aggressivität bei primär erhaltener Intelligenz und Orientierung.
→ 5) Weitere Symptome: Können u.a. Fresssucht, Sammeln von Gegenständen, Harn- und Stuhlinkontinenz sein.
→ 6) Im weiteren Krankheitsverlauf entwickeln sich eine anamnestische Aphasie mit stereotyper Sprache, Palilalie sowie Echolalie und weitere Gedächtnisstörungen.
→ 7) Neurologische Symptome: Mit Rigor, Tremor, Akinese, aber auch Apraxie und Agnosie bis hin zum Auftreten von Primitivreflexen (z.B. Greif-, Saug-, Palmomentalreflex).
→ 8) Das Endstadium ist schließlich gekennzeichnet durch das Vollbild der Demenz.
→ Klinisch-relevant: Bei der frontotemporalen Demenz werden im Hinblick auf die pathologisch-anatomischen Veränderungen un die klinische Symptomatik nochmals zwischen 2 Subtypen differenziert:
→ A) Orbital/Basaltyp: Ist geprägt durch die klinische Symptomatik mit psychomotorischer Unruhe, Enthemmung, Missachtung von Normen und Gesetzen, Unzuverlässigkeit, Taktlosigkeit, sexueller Anzüglichkeit, Witzelsucht etc.
→ B) Konvexitätstyp: Hier stehen völlige Antriebslosigkeit, Sprachverarmung mit z.T. Persevation und/oder Echolalie sowie Apathie im Vordergrund.
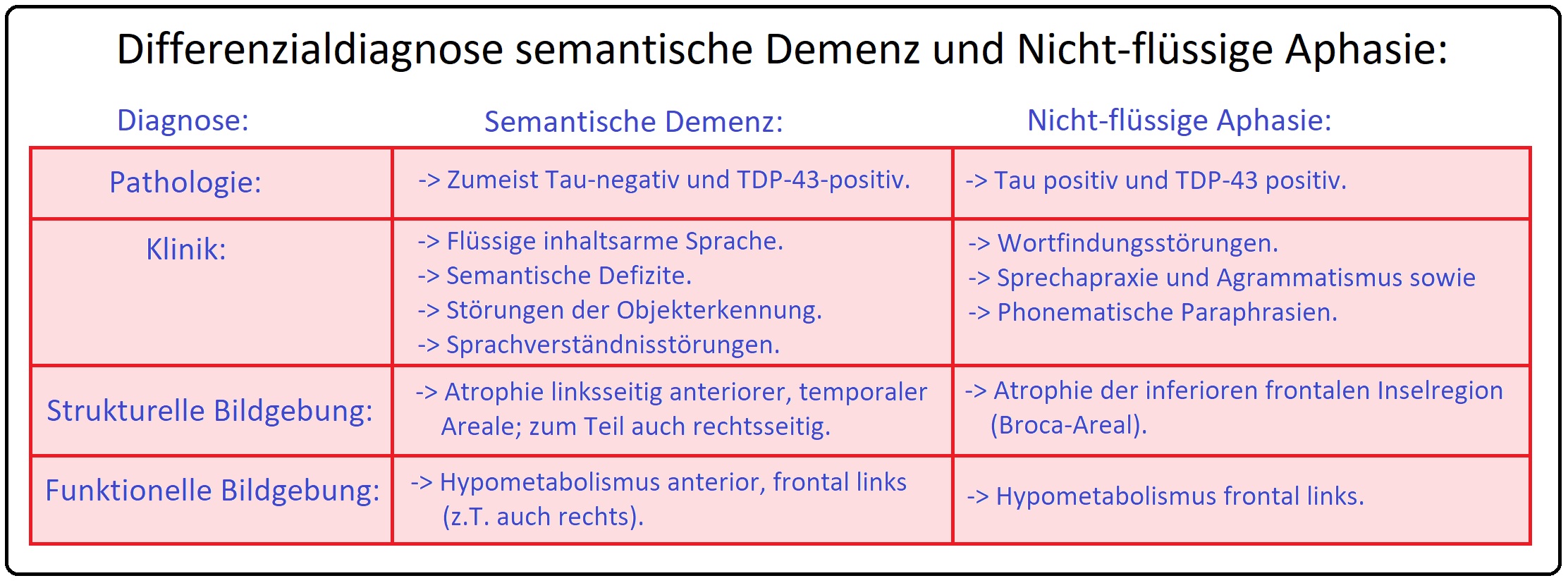
→ II: Semantische Demenz/Aphasie: Entwickelt sich infolge einer Degeneration insbesondere des linken Temporallappens. Sie wird zu Krankheitsbeginn und im weiteren Verlauf durch Störungen des Bedeutungsinhaltes (= Semantik) charakterisiert. Des Weiteren manifestieren sich frühzeitig sowohl eine semantische Aphasie (= gestörtes Verständnis des Sinns von Wörtern) als auch assoziative Agnosie (= gestörtes Wissen um Objekte). Klinisches Korrelat stellen Benennungsstörungen und semantische Paraphrasien dar. Schließlich geht die semantische Aphasie in das Vollbild der Demenz über.

→ III: Progressive nicht-flüssige Aphasie: Bei dieser Variante ist insbesondere die linke Präfrontalkortex im Broca-Areal betroffen. Sie ist durch eine nicht flüssige Sprache mit konsekutiven Wortfindungsstörungen und langen Pausen (ähnlich der Broca-Aphasie) charakterisiert, wobei bei den Betroffenen die Persönlichkeit lange Zeit intakt ist und zu einem erheblichen Leidensdruck führt. Der Verlust der Sprache (mit Symptomen wie Agrammatismus bzw. Telegrammstil, phonematische Paraphrasie, Sprechapraxie, etc.) schreitet unaufhaltsam fort bis das Ausdrucksvermögen vollständig schwindet und der Patient mutistisch wird. In diesem Stadium manifestieren sich zusätzlich noch Gedächtnis- und Störungen der visuellen und räumlichen Funktion.
→ Diagnose:
→ I: Anamnese/Klinische Untersuchung:
→ 1) Hierbei spielt gerade die Fremdanamnese eine bedeutende Rolle zur Abklärung einer positiven Familienanamnese (Verwandte 1. Grades), Krankheitsbeginn (vor dem 60. Lebensjahr), etc.
→ 2) Klinisch lassen sich evtl. Faszikulationen, Muskelschwäche, -schwund und Primitivreflexe nachweisen.
→ 3) Psychologische Testverfahren zur Erfassung von Verhaltensauffälligkeiten z.B. Frontal-Behavioral-Inventory (= FBI).
→ Klinisch-relevant:
→ A) Beim FBI handelt sich um einen Fremdanamnesebogen mit insgesamt 24 Items, die von 0 (keine) bis 4 (meist/schwere) bewertet werden.
→ B) Die Items umfassen u.a. nachfolgende Themen der Persönlichkeit wie Aspontanität, Affekt, Verlust der Einsicht, Vernachlässigung, Perseveration, Witzeln, Impulsivität, Hyperoralität, Hypersexualität, Inkontinenz, etc.
→ C) Eine Gesamtsummenscore von > 30 Punkten bestätigt eine Frontallappenläsion und kann u.a. zur Abgrenzung gegenüber der Alzheimer-Demenz eingesetzt werden.
→ II: Bildgebende Verfahren:
→ 1) cCT/cMRT: Atrophie der Frontal- und/oder der Temporallappen im konsekutiver Erweiterung des äußeren Liquorraumes.
→ 2) SPECT/PET: Minderperfusion und Hypometabolismus (Glukosestoffwechselstörungen) in den frontotemporalen Arealen.
→ III: Liquoruntersuchung: Evtl. Nachweis des Tau-Proteins im Liquor.
→ IV: Histopathologie: Histologisch sind u.a. Nervenzellverluste, eine Gliose sowie zytoplasmatische Tau-positive oder Ubiquitin-positive Einschlusskörper nachweisbar.
→ V: EEG: Patienten mit frontotemporaler Demenz zeigen sehr lange ein unauffälliges EEG in Kontrast zu dem schweren klinischen Krankheitsbild.
→ Differenzialdiagnose: Hiervon z.T. sehr schwierig abzugrenzen sind u.a.:
→ I: Alzheimer-Demenz: Hierbei manifestieren sich die kognitiven Defizite deutlich frühzeitiger als die Wesens-/Verhaltensveränderungen.
→ II: Vaskuläre Demenz: Eruierung von metabolischen Risikofaktoren, evtl. ischämische Insulte in der Vorgeschichte, etc.
→ III: Lewy-Körper-Demenz: Nachweis einer Parkinson-Symptomatik.
→ IV: Auch im Rahme einer Creuzfeldt-Jakob-Krankheit können frontotemporale Hirnareale mitbetroffen sein.
→ Therapie:
→ I: Allgemein:
→ 1) Eine kausale Therapie existiert noch nicht; es wird jedoch eine gut strukturierte Umgebung für den Betroffenen empfohlen.
→ 2) Primär sollte eine umfangreiche Aufklärung der Angehörigen hinsichtlich der zunehmenden Beaufsichtigung und Betreuung erfolgen.
→ 3) Bei Patienten mit progredienter nicht-flüssiger Aphasie ist eine adäquate Logopedie obligat.
→ II: Medikamentöse Therapie:
→ 1) Insbesondere bei Antriebs- und Affektstörungen mit Depressivität haben sich Antidepressiva gerade vom SSRI-Typ (z.B. Paroxetin) mit/ohne Carbamazepin (oder Valproat) etabliert.
→ 2) Atypische Neuroleptika in geringer Dosis habe einen guten Effekt auf die "Positivsymptomatik" wie z.B. Agitiertheit, Logorrhoe und Aggressivität, ohne die Kognitionen zu beeinträchtigen.
→ 3) Besteht ein hypersexuelles Verhalten kann ein Therapieversuch Sertralin erwogen werden.
→ 4) Manifestieren sich im weiteren Krankheitsverlauf schließlich kognitive Defizite wird eine Therapie mit Nootropika (z.B. Cholinesterasehemmer, Glutamat-Modulatoren) empfohlen.
→ Prognose:
→ I: Die frontotemporale Demenz weist einen progredienten Krankheitsverlauf auf, die im Endstadium zu Bettlägerigkeit und völliger Pflegebedürftigkeit führt.
→ II: Die Erkrankung endet meist nach 7-10 Jahren infolge einer Pneumonie und/oder den Folgen einer Kachexie letal.
- Details
- Geschrieben von: CF
- Kategorie: Organische psychische Störungen
- Zugriffe: 13265
→ Definition:
→ I: Bei der vaskulären Demenz handelt es sich um eine heterogene Gruppe von demenziellen Syndromen, die auf eine zerebrale Durchblutungsstörung zurückzuführen ist.
→ II: Hierbei spielt der zeitliche Zusammenhang zwischen der demenziellen Symptomatik und der vaskulär bedingten Hirnläsion mit ihren neurologischen Symptomen eine wichtige Rolle.
→ Epidemiologie:
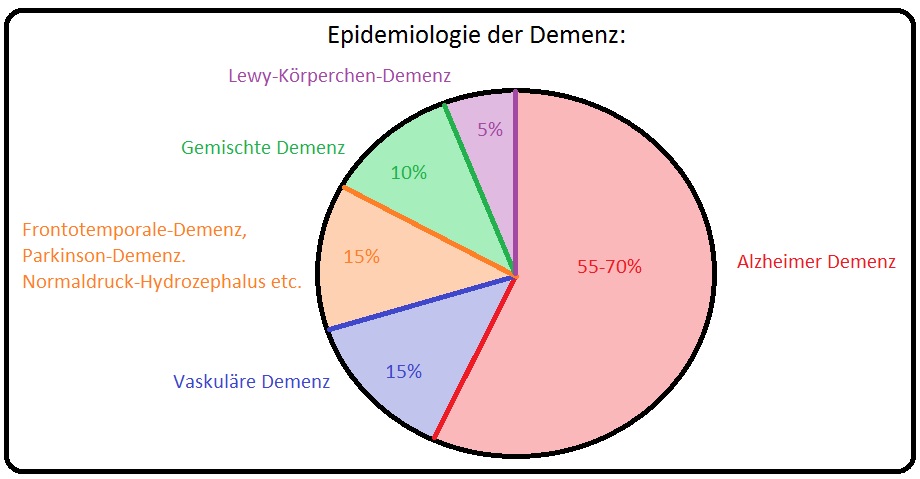
→ I: Die Inzidenz für das Auftreten einer vaskulären Demenz liegt in Deutschland bei den > 65. Jährigen bei bis zu 7% und stellt nach der Alzheimer-Krankheit die 2-häufigste Demenzform dar.
→ II: Der Manifestationsgipfel liegt zwischen dem 60.-70. Lebensjahr, wobei Männer deutlich häufiger betroffen sind als Frauen.
→ III: In 10% der Fälle handelt sich um eine Mischformen zwischen vaskulärer - und Alzheimer-Demenz.
→ Ätiologie: Die Genese dieser Demenz-Form beruht auf vaskulären Erkrankungen, die zu einer disseminierten Hirnparenchymschädigung, in deren Folge es zur Beeinträchtigung intellektueller Funktionen kommt. Ursachen sind vor allem:
→ I: Arteriosklerose:
→ 1) Arteriosklerotische Gefäßveränderungen im Rahmen von Mikro- (keiner Arterien mit lakunärer, subkortialer Lokalisation) und/oder Makroangiopathien (die zu kortikalen Territorial- und Grenzzoneninfarkten führen).
→ 2) Arteriosklerose der Hirngefäße mit sekundärer Degeneration des Hirnparenchyms.
→ II: Weitere Ursachen: Sind eher selten; hierzu zählen:
→ 1) Thrombosen und Embolien.
→ 2) Amyloidangiopathie,
→ 3) Blutgerinnungsstörungen,
→ 4) Vaskulitiden etc.
→ III: Risikofaktoren: Sind u.a. arterielle Hypertonie (insbesondere beim Morbus Binswanger), Diabetes mellitus, Hyperurikämie, Hyperlipidämie, insbesondere die Hypercholesterinämie, Gerinnungsstörungen, Adipositas, Bewegungsmangel, Nikotinabusus etc.
→ Klassifikation: Nach ICD-10 wird die heterogene Gruppe der vaskulären Demenz in nachfolgende Subtypen unterteilt:
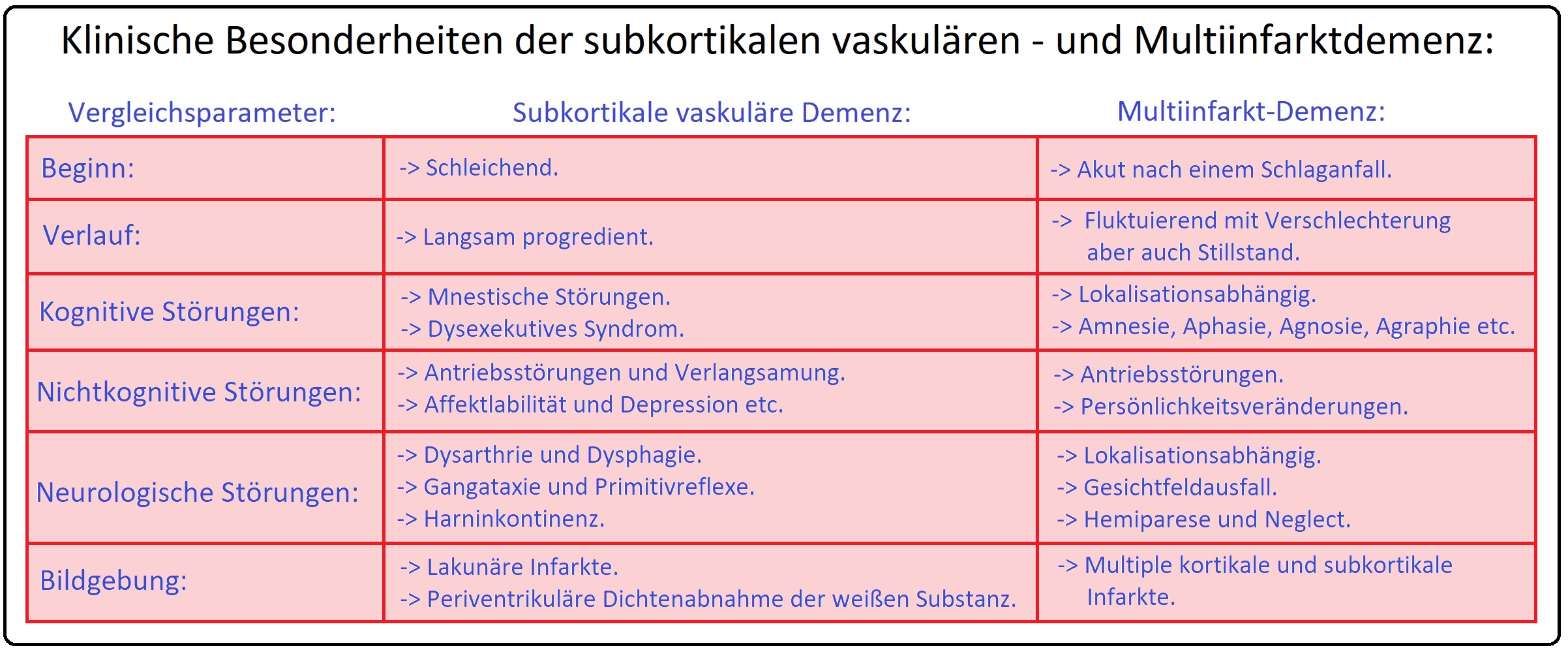
→ I: Multiinfarkt-Demenz: Sie beginnt zumeist einschleichend nach mehreren kleinen ischämischen Ereignissen, die in der Folge zur Anhäufung von multiplen lakunären Hirnparenchymläsionen bzw. kortikalen Territorialinfarkten führen:
→ 1) Kortikale Territorialinfarkte: Ursache hierbei sind thromboembolische Verschlüsse zumeist im Stromgebiet der Arteria cerebri media oder posterior.
→ 2) Subkortikale lakunäre Demenz: Betrifft die kleineren und kleinsten Arterien (= Mikroangiopathie) und weist typische Prädilektionsstellen in Stammganglien, Hirnstamm und der Capsula interna auf.
→ II: Vaskuläre Demenz mit akutem Beginn: Diese Form entwickelt sich rapide, typischerweise nach vorangegangenen apoplektischen Insulten, aber auch infolge von zerebrovaskulären Thrombosen und Embolien, seltener nach massiven Blutungen.
→ Klinisch-relevant: Zudem existieren sogenannte strategische Infarkte, die zumeist nur eine geringe Ausdehnung aufweisen, aber bei bilaterale Lokalisation an strategisch wichtigen Stellen wie z.B. der Thalamus oder Hippocampus schwere Störungen hervorrufen können.
→ III: Demyeliniserung des Marklagers: (= Morbus Binswanger = subkortikale vaskuläre Demenz)
→ 1) Der Morbus Binswanger ist gekennzeichnet durch das Auftreten von ausgedehnten, zumeist periventrikulär und okzipital gelegenen Marklagerläsionen und Aussparung der Kortex.
→ 2) Histopathologisch zeigt sich eine massive Demyelinisierung der weißen Substanz, lakunäre Infarkte sowie eine deutliche Erweiterung der Seitenventrikel.
→ 3) Ursachen sind fibrohyaline Umbauprozesse und Sklerosierung der Markarteriolen (arteriosklerotisch) aufgrund einer langjährig bestehenden arteriellen Hypertonie (die Hirnrinde bleibt ausgespart).
→ 4) Die Binswanger-Krankheit tritt jenseits des 50. Lebensjahr auf und schreitet schleichend und zumeist langsam fort.
→ Klinisch-relevant: Das klinische Bild des Morbus Binswanger ist geprägt durch:
→ A) Einen kontinuierlichen kognitiven Leistungsverlust.
→ B) Neurologische Symptome: Mit Hypokinesen, Pseudobulbärparalysen, Gangataxie und Harninkontinenz sowie
→ C) Psychische Störungen: Häufig manifestieren sich depressive Verstimmungen und Persönlichkeitsveränderungen mit Antriebsstörungen, aber auch emotionale Labilität.
→ IV: Gemischte vaskuläre Demenzen: Hierunter fallen die häufig auftretenden Mischformen der vaskulären Demenz und Alzheimer-Krankheit. Sie sind sowohl kortikal als auch subkortikal lokalisiert.
→ Klinik: Vaskuläre Demenzen weisen aufgrund der sehr unterschiedlichen Ursachen und Pathomechanismen eine uneinheitliche klinische Symptomatik (z.B. z.T. plötzlicher Krankheitsbeginn, aber auch schleichende Verlaufsformen sind möglich) auf, sodass alle Symptome einer Demenz auftreten können. Hierzu zählen:
→ I: Persönlichkeitsveränderungen: Erhöhte Reizbarkeit, Aggressivität, aber auch emotionale Labilität, Antriebs- und Interessenlosigkeit, sozialer Rückzug, Apathie (zumeist ausgeprägter als bei der Alzheimer-Demenz).
→ II: Kognitive Störungen: Mit Gedächtnisstörungen, Störungen der Konzentration und der Auffassung und nicht zuletzt Orientierungs- und Sprachstörungen, Apraxie und Agnosie.
→ III: Insbesondere bei der Multiinfarktdemenz können sich Schlafstörungen mit nächtlichen Verwirrtheitszuständen und paranoid-halluzinatorischen Epidosen entwickeln.
→ IV: Weitere Symptome: Sind u.a. Affektlabilität mit vorübergehender depressiver Stimmung, aber auch unbeherrschtem Lachen, Weinen sowie deliranten Episoden.
→ V: Zusätzlich und diagnostisch richtungsweisend sind neurologische Ausfälle aufgrund von vaskulären Pathomechanismen. Hierzu zählen insbesondere:
→ 1) Rezidivierende Stürze,
→ 2) Motorische und sensible Ausfälle,
→ 3) Hemiparesen,
→ 4) Positive Pyramidenbahnzeichen,
→ 5) Kleinhirnsymptome etc.
→ Klinisch-relevant: Bei den betroffenen Patienten manifestieren sich häufig weitere arteriosklerotisch bedingte Erkrankungen wie koronare Herzkrankheit, periphere arterielle Verschlusskrankheit, Z.n. Myokardinfarkt etc.
→ Verlaufsformen: Bei der vaskulären Demenz existieren vor allem 2 wichtige Krankheitsverläufe:
→ I: Allmählicher Beginn: Hervorgerufen durch eine Mikroangiopathie mit eher schleichend progredientem Beginn und Verlauf. Sie beginnt zumeist mit Müdigkeit, Kopfdruck, Schwindel, Unruhe, Schlafstörungen, im weiteren Krankheitsverlauf zeigen sich kognitiven Beeinträchtigungen mit Merk- und Konzentrationsstörungen sowie Vergesslichkeit.
→ II: Plötzlicher Beginn: Verursacht durch eine Makroangiopathie infolge einer TIA, PRIND oder eines apoplektischen Insultes; der Krankheitsverlauf ist schubweise. Typische klinische Zeichen sind Verwirrtheitszustände sowie akute neurologische Ausfälle (Sensibilitätsstörungen, Gangunsicherheit, Herdzeichen, Paresen, weitere fokal-neurologische Symptome etc.).
→ III: Ein weiteres Charakteristikum der vaskulären Demenz ist, dass sich im Krankheitsverlauf rezidivierende, vorübergehende aber auch wechselnde neurologische Herdsymptome eruieren lassen.
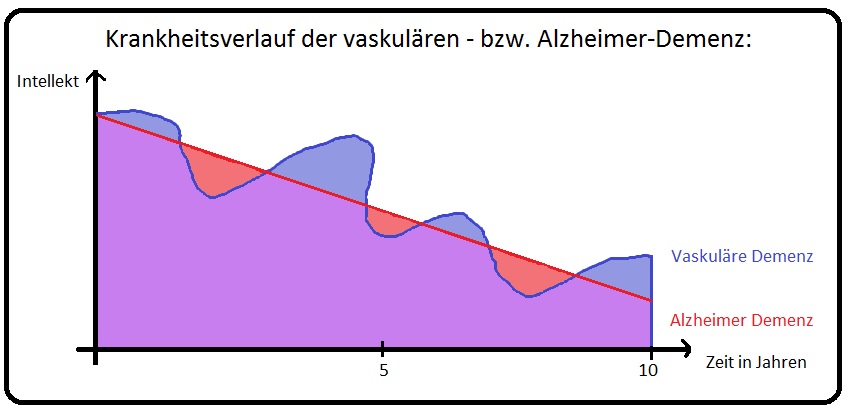
→ Diagnose: Wichtig für die Diagnose ist die Feststellung eines demenziellen Syndroms sowie der Nachweis einer zerebrovaskulären Erkrankung und das Erbringen eines kausalen Zusammenhangs. Eine endgültige Diagnosebestätigung ist erst post mortem durch eine histopathologische Untersuchung des Gehirns möglich.
→ I: Anamnese/klinische Untersuchung: Hierbei steht Risikofaktoren, Vorerkrankungen, Erkrankungsbeginn und -verlauf (Eigen-/Fremdanamnese) im Vordergrund. Klinische Eruierung des Neurostatus (insbesondere neurologische Herdbefunde), aber auch des internistischen Status mit Abklärung des Gefäßstatus (pAVK, KHK, Fundus hypertonicus etc.).
→ II: Labor:
→ 1) Mit Bestimmung des Routinelabors (CRP, BSG, Elektrolyten, Gerinnungsparameter Quick, pTT, Leber-, Pankreasenzyme, Nierenretentionswerte, sowie LDL, Vitamin B1, B12 etc.).
→ 2) Liquordiagnostik: Zum Ausschluss von entzündlichen Erkrankungen des ZNS.
→ III: Bildgebende Verfahren:
→ 1) Dopplersonographie: (der hirnversorgenden Gefäße) Zur Darstellung von möglichen Plaques, Stenosen und Gefäßverschlüssen.
→ 2) Herzecho: Bei unklarem Befund und z.B. stattgefundener Endokarditis.
→ 3) cCT/cMRT: Zum Nachweis von hypodensen Infarktbereichen, lakunäre Infarkte und ausgedehnte Demyelinisierungen vorwiegend im Marklager und den Großhirnhemisphären.
→ 4) SPECT: (= Singel-Photon-Emission-Computer-Tomographie) Darstellung möglicher Perfusionsstörungen.
→ IV: Neuropsychologische Testverfahren: Zur Eruierung von objektiven kognitiven Leistungseinbußen. Hierzu gehören u.a. der Demenz-Test, Minimal-Mental-Test oder die Hachinski-Ischämie-Skala. Sie dient der klinischen Unterscheidung zwischen vaskulärer und degenerativer Demenz und beinhaltet nachfolgende Parameter.
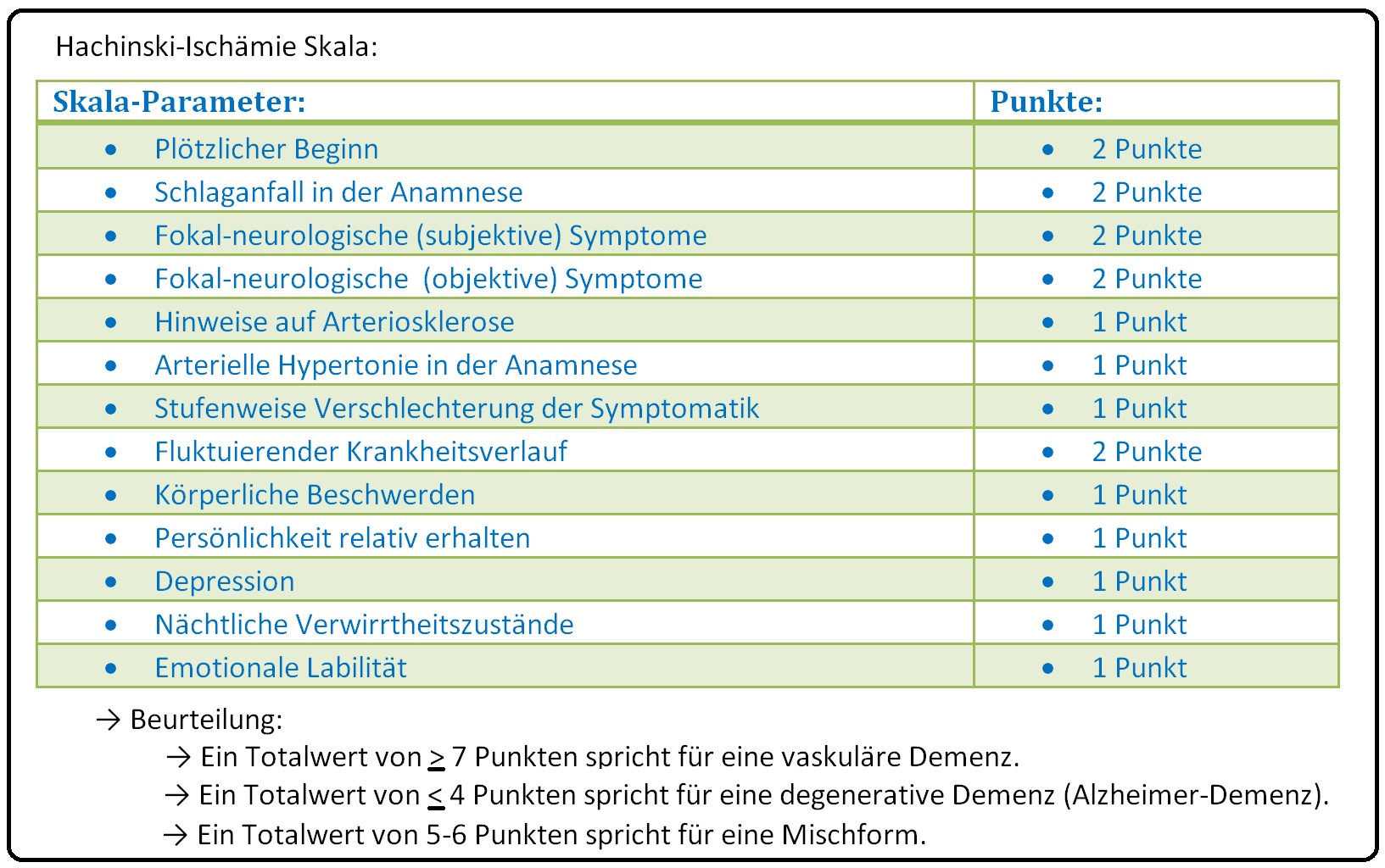
→ Differenzialdiagnose: Von der vaskulären Demenz müssen insbesondere nachfolgende Erkrankungen abgegrenzt werden; hierzu zählen:
→ I: Weitere demenzielle Syndrome wie die:
→ 1) Alzheimer-Demenz,
→ 2) Frontotemporale Demenz,
→ 3) Lewy-Body-Demenz,
→ 4) Demenz bei der Parkinson-Krankheit.
→ 5) Normaldruckhydrozephalus.
→ II: Pseudodemenz bei Depression,
→ III: Der manchmal auftretende Wahn lässt evtl. an eine Erkrankung aus dem schizophrenen Formenkreis (z.B. Schizophrenie) denken.
→ Therapie: Die vaskuläre Demenz kann durch frühzeitige Behandlung der Grunderkrankung bzw. der Risikofaktoren verhindert werden.
→ I: Behandlung der Risikofaktoren: Die Reduktion bzw. Beseitigung der Risikofaktoren (für die Entwicklung eines Schlaganfalls) ist eine präventive Maßnahme und beinhaltet u.a. die frühzeitige Behandlung einer arteriellen Hypertonie und eines bestehenden Vorhofflimmerns durch Antihypertensiva bzw. Antikoagulanzien (z.B. ASS, Macumar).
→ Klinisch-relevant: Der arterielle Blutdruck darf nicht zu schnell gesenkt werden, um keine zerebrale Minderperfusion auszulösen.
Auch eine gesunde Lebensweise (Ernährung, Bewegung etc.) dient der Reduktion von Risikofaktoren.
→ II: Therapie kognitiver Symptome: Zur Beeinflussung des zellulären Sauerstoff und Glukosestoffwechsels haben sich Nootropika wie die Acetylcholinesterase-Hemmer (z.B. Rivastigmin 3-6mg/d) oder die Glutamat-Modulatoren (z.B. Memantin 10-20mg/d) etabliert.
→ III: Sekundäre Krankheitssymptome: Wie Unruhe, Depression, Angst aber auch wahnhafte Symptome sollten mit schwachpotenten, dämpfenden Antipsychotika behandelt werden. Wahnhafte Symptome sind aufgrund des veränderten zerebralen Stoffwechsels und der weiteren zumeist bestehenden Komorbiditäten nur schwer zu therapieren. Von den atypische Neuroleptika wie z.B. Risperidon oder Olanzapin ist aufgrund der erhöhten Mortalität abzuraten, vielmehr kann die Applikation eines klassischen Neuroleptikums wie z.B. Haloperidol in einer sehr niedrigen Dosis (0,5mg/d) versucht werden. Alternativ ist die Gabe von Clozapin (12,5-75mg/d) oder Quetiapin (25-100mg/d) zu erwägen.
→ Klinisch-relevant: Die Benzodiazepine sind wegen der möglichen paradoxen Reaktion relativ kontraindiziert.
→ IV: Weitere Interventionen: Supportive Behandlungsmaßnahmen sind bei der vaskulären Demenz u.a.:
→ 1) Kognitives Training mit Wahrnehmungs-, Konzentrations- und Gedächtnisübungen.
→ 2) Bewegungsübungen und Sport
→ 3) Aufrechterhaltung und Förderung von Hobbys wie Lesen, Radiohören, etc. und nicht zuletzt die
→ 4) Beratung von Bezugspersonen.
→ Prognose: Die vaskuläre Demenz weist zumeist einen wechselhaft progredienten Verlauf auf und ist schwer vorhersehbar. Erfolgt eine frühzeitige und adäquate Therapie der Risikofaktoren wie arterielle Hypertonie, Herzrhythmusstörungen, Diabetes mellitus etc. ist die Prognose der zerebrovaskulären Demenz insgesamt günstiger als bei den primär-degenerativen Demenzen (Alzheimer-Typ).
- Details
- Kategorie: Organische psychische Störungen
- Zugriffe: 17261
→ Definition: Bei der Alzheimer Demenz handelt es sich um eine kognitive Leistungsstörung aufgrund einer neurofibrillären Degeneration der Nervenzellen durch Ablagerung von extrazellulären kortikalen Plaques.
→ Klassifikation: Diese erfolgt nach der Altersmanifestation:
→ I: Alzheimer-Demenz mit frühen Beginn vor dem 65. Lj. (= präsenil).
→ II: Alzheimer Demenz mit spätem Beginn nach dem 65. Lj. (= senil).
→ Epidemiologie: Derzeit gibt es in Deutschland 1 Million diagnostizierte Demenzerkrankte, bei denen in 70% der Fälle eine Alzheimer-Erkrankung vorliegt. Mit einer Prävalenz von 5% ist es die häufigste Ursache eines kognitiven Defizites bei Patienten > 65. Lebensjahr.
→ Ätiologie: Hierbei werden 2 Formen unterschieden:
→ I: Sporadisch auftretende Form: Ursache nicht bekannt, mit 95% die häufigste Form (Zwillingsstudien belegen jedoch eine genetische Komponente).
→ II: Familiär auftretende Form: Hierbei handelt es sich um einen autosomal-dominanten Erbgang mit Mutationen im APP-Gen (= Amyloid-(Vorläufer)-Präkusor-Protein), bzw. in den Presinilin-Genen PS-1/PS-2 auf den Chromosomen 1,14 und 21; es besteht eine vermehrte Häufigkeit bei Verwandten 1. Grades (findet man in 3% der Fälle).
→ Risikofaktoren: Die eine Alzheimer-Demenz begünstigen:
→ I: Das Alter: Ist ein bedeutender Aspekt. Hierbei liegt das Risiko bei der Altersklasse zwischen 30-59 Jahren bei 0,02%, zwischen 60-69 Jahren bei 0,3%, zwischen 70-79 Jahren bei 3,2% und bei 80.-90.-Jährigen schon bei 10,8%.
→ II: Neurologische Erkrankungen: Bei Verwandten 1. Grades mit demenziellen Syndrom ist das Risiko erhöht. Eine weitere Ursachen ist ein Schädel-Hirn-Trauma in der Vorgeschichte.
→ III: Genetische Aspekte: Das Tragen des Apolipoprotein-E4-Allels (APO-E4) erhöht das Risiko einer Demenz. Bei Heterozygoten um das 3-fache, bei Homozygoten um das 10-fache.
→ IV: Weitere Risikofaktoren: Sind arterielle Hypertonie, Hypercholesterinämie, Diabetes mellitus, Adipositas, körperliche Inaktivität, aber auch geringe Schuldbildung und Intelligenz.
→ V: Protektive Faktoren: Hierzu gehören die rheumatoide Arthritis, die jahrelange Einnahme von NSAR, Östrogen und Statinen.
→ Pathogenese: Die Pathologie der Alzheimer Demenz wird durch intra-/extrazelluläre Ablagerungen (Plaques) bzw. Veränderungen hervorgerufen; Sie bstehen aus:
→ I: Amyloid-ß-Peptid (= extrazelluläre amyloide Plaques) sowie
→ II: Intrazelluläre Neurofibirillenveränderungen, sogenannte Tangles, die sich aus hyperphosphorylierten Tau-Proteinen zusammensetzen.
→ Klinisch-relevant: Es kommt gerade im Bereich des Hippocampus, des basalen Vorderhirns und im Bereich des Temporallappens zur Degeneration mit einer typischen Symptomkonstellation:
→ A) Gedächtnisstörungen,
→ B) Visuelle-räumliche Störungen und
→ C) Benennungsstörungen.
→ Biochemie: Bei der Spaltung des Amyloid-Vorläuferproteins unterschiedet man 2 Wege:
→ I: Nicht-amyloidogener Weg: Hierbei wird das APP durch die Alpha-Sekretase innerhalb des Amyloid-ß-Peptids gespalten. Es entsteht ein Produkt namens C83. Dieses wird durch die y-Sekretase weiter gespalten, wobei kein ß-Amyloid-Peptid entsteht. Vielmehr entsteht ein lösliches Proteinfragment APPs-Alpha, welches wachstumsfördernd und protektiv auf die Nervenzellen wirkt.
→ II: Amyloidogene Weg: APP wird durch die ß-Sekretase gespalten. Es entsteht C99, welches wiederum durch den y-Sekreatase-Komplex gespalten wird. Es entstehen die ß-Amyloid-Peptide. Diese bilden toxische Oligomere, die sich in Form von Plaques zusammenlagern.
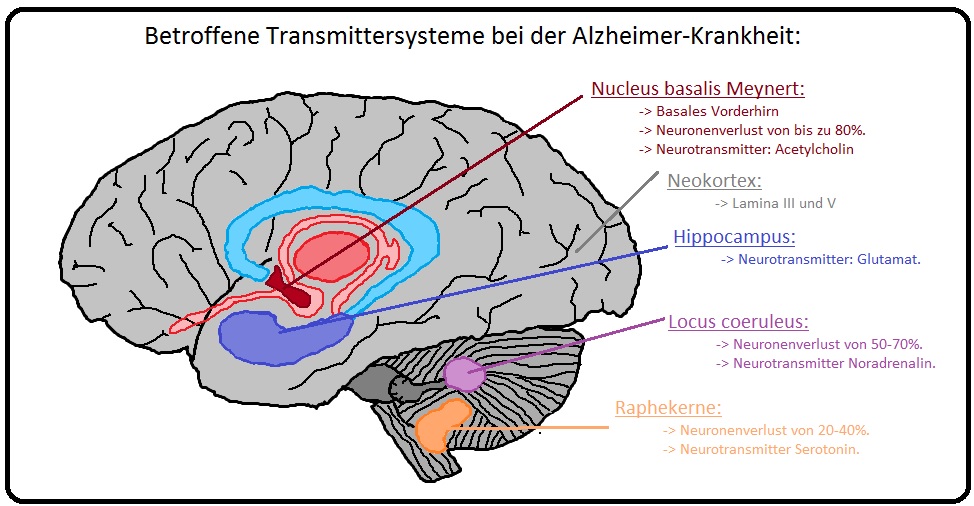
→ Klinisch relevant: Betroffene Transmittersysteme bei der Alzheimer Demenz sind insbesondere:
→ A) Cholinerge Transmitter: Verminderung der Acetylcholintransferase und des Acetylcholins im Bereich des Nucleus balsalis Meynert und anderer Strukturen des balsalen Vorderhirns.
→ B) Folgen: Sind vor allem:
→ 1) Es kommt zu einem Neuronenverlust von bis zu 80%.
→ 2) Beeinträchtigung der Aufmerksamkeit und des Gedächtnisses.
→ 3) Das cholinerge Defizit im Gehirn korreliert mit dem Schweregrad der Demenz und dem Ausmaß der Ablagerung von Amyloidplaques.
→ C) Das cholinerge Defizit wird symptomatisch mittels Cholinesterase-Hemmern behandelt.
→ D) Weitere Transmittersysteme: Sind u.a.:
→ 1) Glutamattransmitter-Sytem: Verlust von Glutamatrezeptoren im Bereich der Neokortex und des Hippocampus. Auch die Reduktion von Glutamatrezeptoren korreliert mit dem Grad der Demenz.
→ 2) Noradrenalintransmitter-System: Gerade im Bereich des Locus coeruleus kann sich ein Neuronenverlust von 50-70% entwickeln.
→ 3) Serotonintransmitter-System: Betrifft die Raphekerne mit einem Neuronenverlust von 20-40%.
→ E) Aber auch inflammatorische Aspekte, vaskuläre Veränderungen spielen bei der Entstehung der Alzheimer Demenz eine wichtige Rolle.
→ Klinik:
→ I: Symptome im Frühstadium:
→ 1) Die Patienten oder Angehörige nehmen als 1. Symptom eine zunehmende Konzentrationsschwäche und Vergesslichkeit wahr (wird z.T. auf das höhere Alter zurückgeführt). Im weiteren Verlauf kommt es zur Abnahme der Gedächtnisleistung und höherer intellektueller Fähigkeiten mit Verminderung der Alltagsaktivität und sozialem Rückzug.
→ 2) Nicht selten wird das klinische Bild von einer Depression (im Alter spricht am von einer Involutionsdepression) begleitet, sodass die Diagnose erschwert wird.
→ II: Symptome im weiteren Verlauf: Hierbei stehen dann die kognitiven Defizite im Vordergrund mit:
→ 1) Beginnend mit Störungen des Kurzzeit-, später dann auch des Langzeitgedächtnisses.
→ 2) Denkstörungen mit Verlangsamung, zähem Gedankengang, Umständlichkeit, inhaltlicher Einengung, etc.
→ 3) Apraxie: Störungen in den willkürlich zielgerichteten Bewegungsabläufen.
→ 4) Semantische Aphasie mit Wortfindungsstörungen, Benennungsstörungen, aber auch abnehmendem Wortschatz, stockender Sprache, später auch Sprachverständnisstörungen.
→ 5) Agnosie: Nichterkennen von Gegenständen und Personen.
→ 6) Alexie (= Verlust der Lesefähigkeit), Agraphie und Akalkulie.
→ 7) Visuokonstruktion-Störungen: Unfähigkeit, komplexe Formen und Muster zu erkennen und sie zu reproduzieren sowie
→ 8) Störungen der räumlichen Orientierung.
→ III: Nicht kognitive Symptome: Zuspitzung von Eigentümlichkeiten (insbesondere Sparsamkeit und Geiz), Antriebslosigkeit, Interessenverlust, Depression, aber auch innere Unruhe und Aggressivität bis hin zu Wahnvorstellungen und Halluzinationen (vor allem optische). Weitere Aufälligkeiten sind u.a. zielloses Umherlaufen, Sortieren und Sammeln sowie Schlafstörungen mit z.T. vollständigem Verlust des Tag-Nacht-Rhythmus.
→ IV: Weitere Störungen:
→ 1) Neurologische Störungen: Mit Urin- und Stuhlinkontinenz.
→ 2) Verhaltensstörungen: Mit depressiver Verstimmung (affektiven Störungen), apathischem Rückzug, Persönlichkeitsveränderungen, innerer Unruhe, Störungen der Impulskontrolle, allgemeine Schlafstörungen insbesondere Störungen des Schlaf-Wach-Rhythmus, sowie Wahnvorstellungen und Halluzinationen.
→ 3) Internistische Begleiterscheinungen: Aufgrund der Bettlägerigkeit steigt insbesondere im fortgeschrittenem Stadium das Risiko für Muskelatrophie, Kontrakturen, Dekubitusulzerationen, aber auch Pneumonien, Thrombosen und Embolien (z.B. tiefe Beinvenenthrombose, Lungenembolie etc.).
→ 4) Neurologische Begleiterscheinungen: Meist erst im fortgeschrittenen Krankheitsverlauf sind Rigor, Gangstörungen, Pyramidenbahnzeichen, Myoklonien und epileptische Anfälle eruierbar.
→ Klassifikation: (der Alzheimer Demenz) Die Krankheit verläuft zumeist langsam und progredient, wobei alle kognitiven Fähigkeiten gleichermaßen betroffen sind.
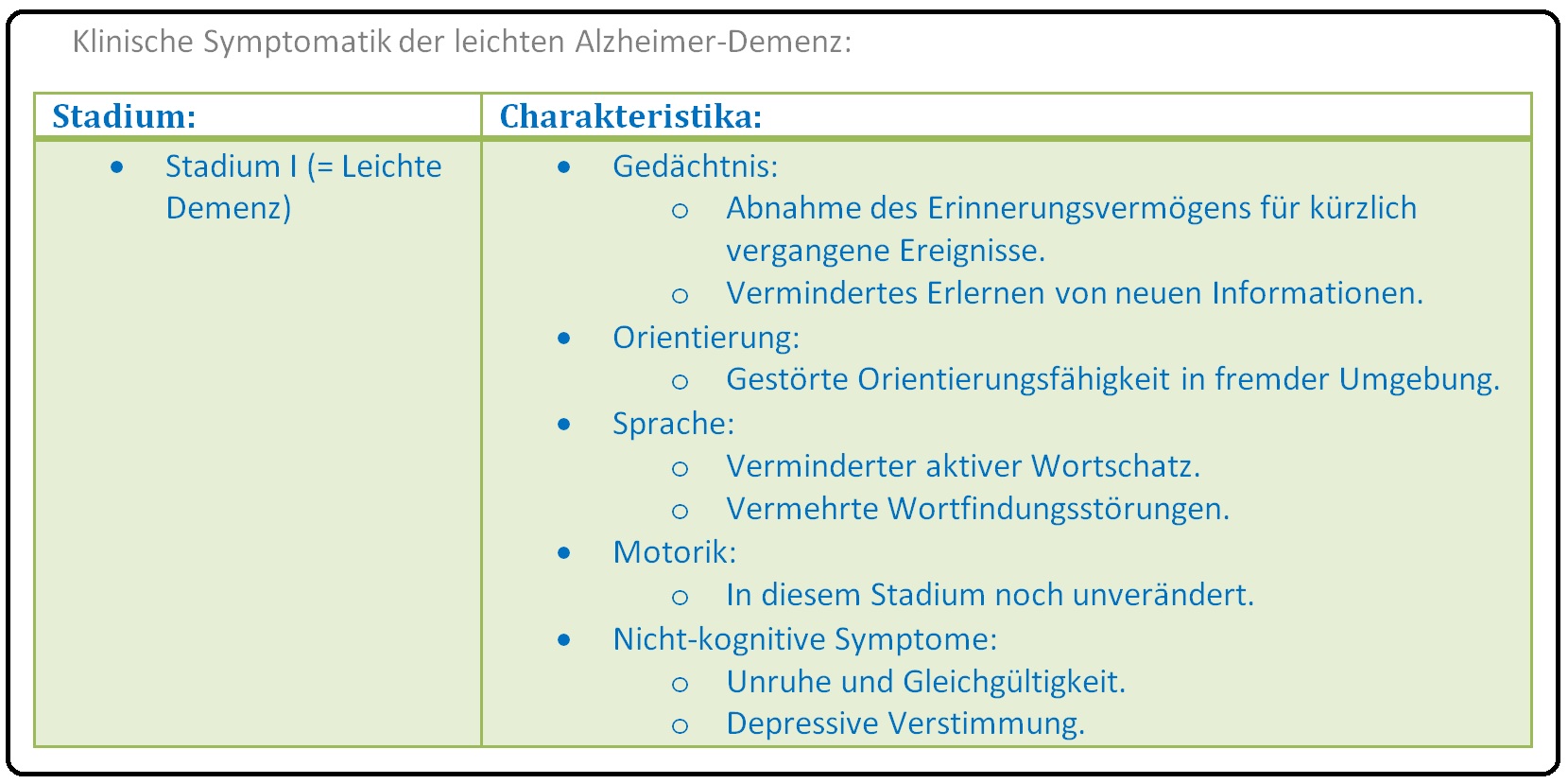
→ I: Stadium I: Leichte Alzheimer Erkrankung:
→ II: Stadium II: Mittelschwere Demenz-Krankheit:
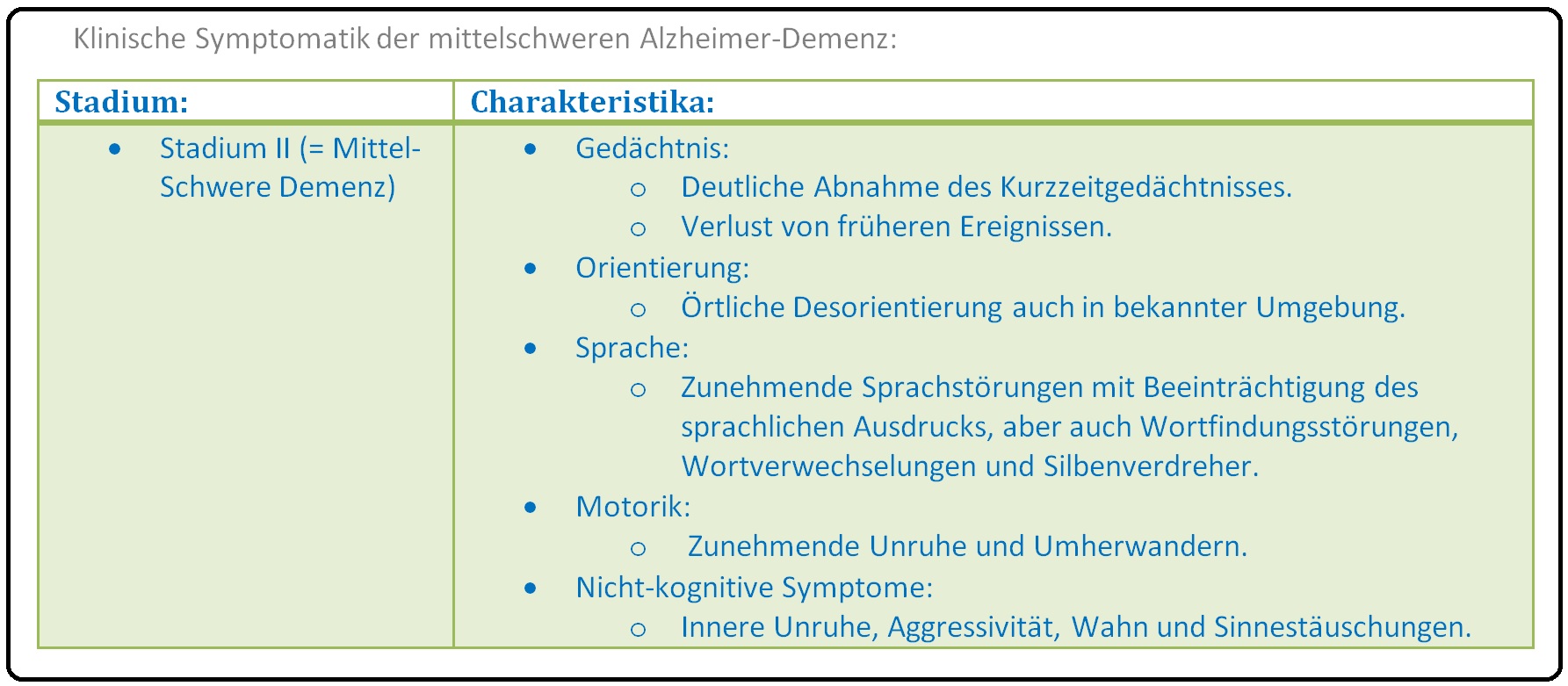
→ III: Stadium III: Schwere Alzheimer-Krankheit:

→ Krankheitsverlauf: Die Verlaufsdauer nach Diagnosestellung der Alzheimer-Demenz beträgt 5-8 Jahre, wobei das Progressionstempo sehr unterschiedlich ist.
→ I: Frühes Stadium: MMST (Minimal-Mental-Status-Test) von 25-20 Punkten; es zeigt sich eine langsame Progression der Erkrankung.
→ II: Mittleres Stadium: MMST 20-10 Punkten; die Progression der Alzheimer-Demenz ist rapide.
→ III: Spätes Stadium: MMST < 10 Punkte; In diesem Stadium ist die Progression wieder langsam, jedoch besteht eine umfassende Pflegebedürftigkeit.
→ Diagnose: Die Alzheimer Demenz ist keine Erkrankung, die man mittels neuroradiologische oder laborchemischer Diagnostik identifiziert, vielmehr wird sie als Ausschlussdiagnose gestellt. Hierbei ist es wichtig, andere Demenzformen, gerade auch die reversiblen Demenzformen, aufgrund von somatischen Erkrankungen auszuschliessen.
→ I: Diagnostische Kriterien: Einer Demenz nach ICD-10:
→ 1) Symptomatik:
→ A) Schleichender Beginn mit langsamer Verschlechterung.
→ B) Fehlen klinischer Hinweise oder spezielle Untersuchungsbefunde, die auf eine andere Demenzform hinweisen.
→ C) Fehlen eines plötzlichen, apoplektischen Insultes oder anderer neurologischer Herdzeichen.
→ 2) Verlaufsform: Derzeit irreversibel.
→ II: Diagnosestellung: Durch die Forschungskriterien nach Dubois:
→ 1) Hauptkriterium: Störung des episodischen Gedächtnis (muss immer vorhanden sein).
→ 2) Nebenkriterien: Hierbei muss mindestens 1 zutreffen.
→ A) Mediotemorale Hirnatrophie durch CT oder MRT nachweisbar.
→ B) Temporoparietaler Hypometabolismus mittels PET nachweisbar.
→ C) Abnahme der ß-Amyloid-Peptid- und Zunahme der Tau-Protein-Konzentration bzw. Zunahme des Gesamt-Tau im Liquor.
→ D) Familiäre Alzheimer Mutation.
→ Klinisch-relevant: Wichtig bei der Diagnosestellung der Alzheimer Demenz ist die Eigen- und Fremdanamnese zum Ausschluss eventueller somatischer Grunderkrankungen oder potentiell-reversibler Demenzformen. Es erfolgt eine klinische Untersuchung zur Erhebung des psychiatrischen, neurologischen und internistischen Status.
→ III: Labor: Hierfür sind noch keine eindeutigen Laborparameter bekannt. Bestimmt werden vor allem:
→ 1) Tau-Protein im Liquor.
→ 2) ß-Amyloid-Peptid (im Frühstadium der Demenz kommt es zu einem Anstieg im fortgeschrittenen Stadium zu einem Abfall des ß-Amyloid-Peptids). Zur Diagnosestellung kann es hinzugezogen werden.
→ 3) Weitere Laboruntersuchungen:
→ A) Blutbild, Blutsenkung, CRP, Elektrolyte einschließlich Ca2+, Blutzucker, Nierenretentionswerte und der Leberwerte.
→ B) TSH basal sowie T3/4;
→ C) Vitamin B12 und Folsäure.
→ D) Coeruloplasmin und Kupfer (zum Ausschluss eines Morbus Wilsons).
→ E) ANA, ANCA, Phospholipid-Antikörper.
→ F) Lues und HIV Serologie
→ IV: Bildgebung: Dient auch nicht der eindeutigen Diagnosestellung, kann jedoch eine Alzheimer Demenz bestätigen.
→ 1) cCT/cMRT: Radiologische Zeichen sind u.a. eine Atrophie des Hippocampus, eine Verbreiterung der Liquorräume (temporoparietal > frontal > occipital). Zudem werden weitere Ursachen wie Normaldruck-Hydrozephalus, Tumoren oder ein subdurales Hämatom ausgeschlossen.
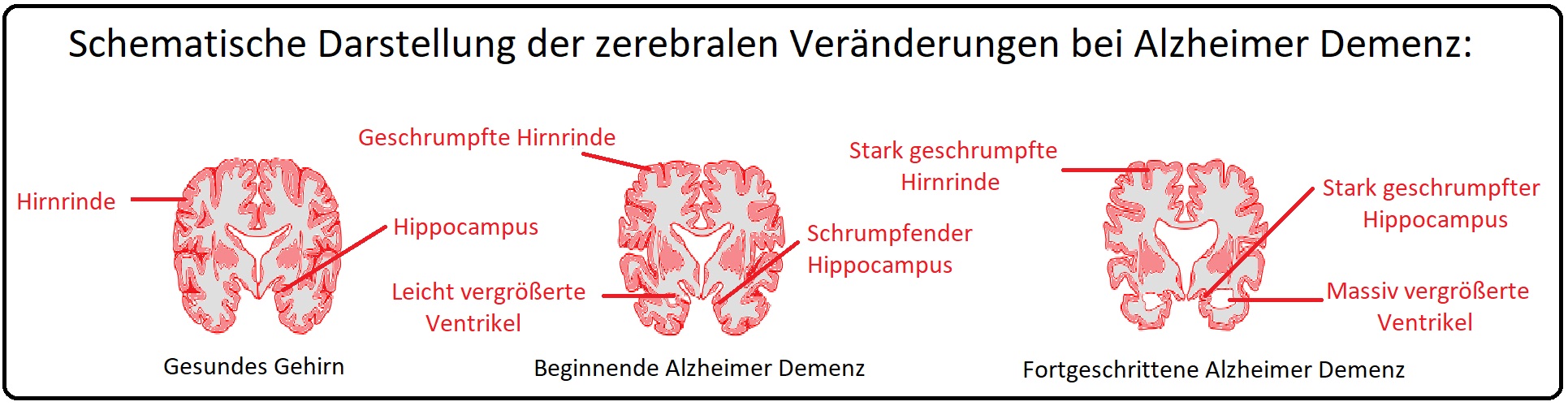
→ 2) PET: Kann die Diagnose Alzheimer Demenz nicht eindeutig gestellt werden, kann ein Positronenemissions-Tomographie mittels 18F-Desoxyglycose hinzugezogen werden. Typischerweise zeigt sich ein Hypometabolismus von Glucose im temporo-parietalen Bereich und frontalen Assoziationsarealen der zerebralen Kortex.
→ V: Zusätzliche Untersuchungen: sind EKG, Herz-Echo und EEG.
→ Differenzialdiagnose: Abgrenzung zu weiteren reversiblen Demenzen, sowie zur vaskulären Demenz.
→ I: Vaskuläre Demenz: Meist bekannte Vorerkrankungen wie Hypercholesterinämie, Adipositas, Diabetes mellitus. Häufig ischämischer Insult in der Vorgeschichte bzw. nachweisbare Läsionen. Klinisch zeigt sich meist ein stufenweiser bzw. durch einen ischämischen Insult hervorgerufener Beginn.
→ II: Des Weiteren müssen auch die anderen demenziellen Syndrome wie:
→ 1) Frontotemporale Demenz,
→ 2) Demenz beim Morbus Parkinson,
→ 3) Lewy-Body-Demenz etc. ausgeschlossen werden.
→ III: Normaldruckhydrozephalus.
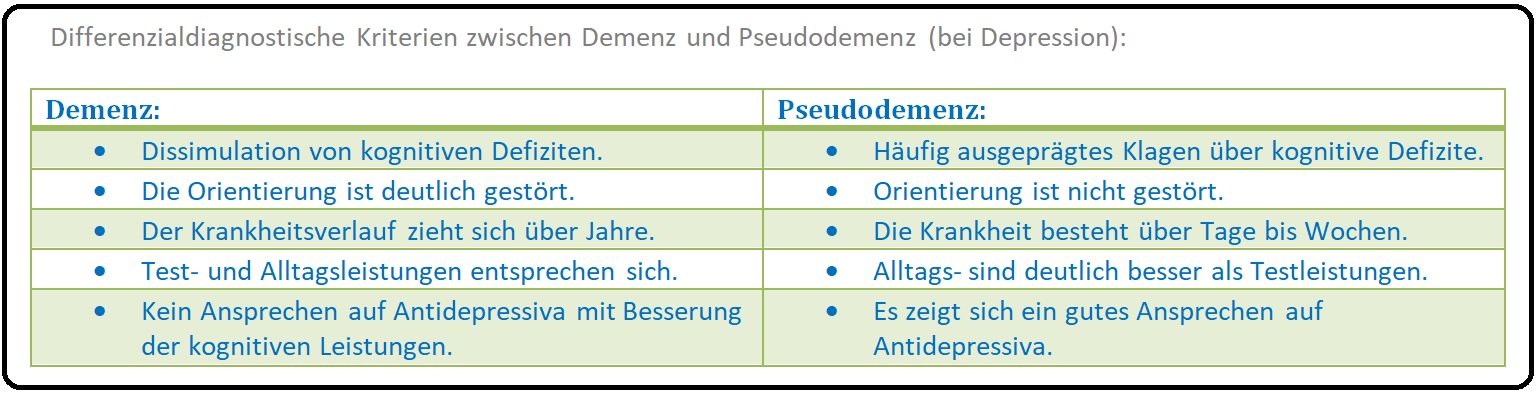
→ IV: Pseudodemenz: (bei Depression) Charakteristisch für das klinische Bild der Alzheimer Krankheit ist u.a., dass der Patient nicht über seine Vergesslichkeit klagt, er den Nachfragen über die kognitive Defizite ausweicht, körperlich nicht krank ist und nicht aus eigenem Antrieb zum Arzt kommt. Für die Differenzialdiagnose, "Demenz-Pseudodemenz" existieren wichtige psychologische Testverfahren wie die Geriatrische Depressions-Skala (GDS) und der Test zur Früherkennung der Demenz mit Depressionsabgrenzung (TFDD).
→ Therapie: Ziel der Therapie ist es, die Erkrankung auf dem Niveau zur Zeit des Therapiebeginns zu halten bzw. eine Progression zu verhindern/verlangsamen. Hierbei ist nicht nur die Verbesserung der kognitiven Leistung zu beachten, sondern auch die Veränderung in der Alltagsaktivität, das klinische Gesamtbild, sowie die Veränderungen des Verhaltens. Besteht eine gute medikamentöse Verträglichkeit sollte diese mindestens für 6 Monate durchgeführt werden und bei klinischer Besserung in eine Langzeittherapie überführt werden. Für die Behandlung der Alzheimer Demenz existieren wichtige Interventionen, zu denen u.a. anchfolgende gehören:
→ I: Medikamentöse Therapie: (Abb.: Dosierungsempfehlungen der Antidementiva bei Morbus Alzheimer) Es sind nur wenige Medikamente (= Antidementiva) vorhanden, die bei ca. 20% der Patienten zu einer Stabilisierung der Symptome für ca. 6-12 Monate, bei gleichzeitigem Fortschreiten des neuropathologischen Befundes, führen. Die früher eingesetzten Nootropika weisen keine Symptomverbesserung auf, sodass sie heute nicht mehr substituiert werden. Einsetzbare Medikamente zur Verbesserung der Demenz-Symptomatik sind heute die:
→ 1) Acetylcholinesterasehemmer: Hierzu gehören Donezepil, Rivastagmin und Galantamin. Sie werden bei leichter bis mittelschwerer Demenz appliziert (Acetylcholinesterasehemmer).

→ 2) Glutamat-Modulatoren: Wie Memantin. Es wird bei mittelschwerer bis schwerer Demenz verabreicht (Glutamat-Modulatoren).

→ II: Weitere Therapiemöglichkeiten: der Demenz-assoziierten Verhaltensstörungen: Mehr als 70% der Patienten weisen im Verlauf der Demenzerkrankung Verhaltensstörungen auf. Hierzu gehören depressive Verstimmung, Angst, Agitiertheit, Aggressionen, Desorientierung, Verwirrung und Wahn.
→ 1) Depression: Bis zu 30% der Patienten sind depressiv. Hierbei können Antidepressiva mit/ohne Kombination mit Antidementiva verabreicht werden. Die trizyklischen AD sollten, aufgrund ihrer anticholinergen Nebenwirkungen (damit haben sie auch einen negativen Einfluss auf die verschiedenen Kognitionen) nicht verabreicht werden, sondern vielmehr die SSRI wie Citalopram oder Sertarlin.
→ 2) Psychotische Symptome: Der Einsatz von Antipsychotika ist bei Wahnvorstellungen wie Eifersuchts- oder Beeinträchtigungswahn, bei Erregungszuständen mit Eigen- oder Fremdverletzung, Desorientierung, Verwirrtheit, Angstzuständen und Schlafstörungen indiziert.
→ A) Hochpotente Antipsychotika: (z.B: Haloperidol) mit einer Initialdosis von 0,5-1mg und einer Höchstdosis von 2-3 mg/d; jedoch aufgrund der extrapyramidalen NW bei älteren Patienten problematisch (klassische Antipsychotika).
→ B) Hochpotente atypische Antipsychotika: Bei schweren, psychotischen Symptomen hat sich gerade Risperidon 0,5-1 mg oder alternativ Aripipralzol (Abilify) etabliert (atypische Antipsychotika).
→ 3) Bei Unruhe- oder Angstzuständen: Sind niederpotente Anticholinergika, mit geringer anticholinerger Wirkung, wie Melperon (25-100mg/d) oder Pipamperon (3x 20-40mg/d) zu verabreichen.
→ Klinisch-relevant: Benzodiazepine sind aufgrund der Beeinträchtigung des Gedächtnisses, ihrer paradoxen Wirkung und der Gefahr der Entwicklung eines Delirs mit Vorsicht einzusetzten.
→ 4) Schlafstörungen: Hierbei können Mirtazapin (7,5-15 mg/d) oder nicht-Benzodiazepin-Hypnotika wie Zolpidem (5-10mg/d) gegeben werden.
→ III: Nicht-medikamentöse Therapie: Hierbei ist es wichtig, den Patienten eine Tagesstruktur aufzuweisen, bei der sie ausreichend mentale und körperliche Aktivität erlangen; dies kann in Tagesstätten erfolgen, gleichzeitig werden Angehörige entlastet. Eine kognitive Therapie hat sich nicht als protektiv erwiesen. Des Weiteren sollten Angehörigen eine ausreichende Psychoedukation, sowie Selbsthilfegruppen angeboten werden.
→ IV: Experimentelle Therapie:
→ 1) Impfung mit ß-Amyloid zur Stimulation der Bildung von Antikörpern, die die Fibrillenaggregation hemmen und konsekutiv die Elimination von toxischen Spaltprodukten steigern.
→ 2) Sekretase-Hemmern: Substitution von Medikamente, die die ß/ySekretase hemmen (diese bildet das toxische ß-Amyloid).
→ 3) Sekretase-Stimulatoren: Stimulieren die Alpha-Sekretase, zur Bildung von protektiven APP-Fragment.