- Details
- Geschrieben von: CF
- Kategorie: Hypophyse
- Zugriffe: 10685
→ Definition: Beim Schwartz-Bartter-Syndrom handelt es sich um eine seltene Erkankung, die durch eine vermehrte inadäquate ADH-Sekretion mit konsekutiver Wasserretention und Verdünnungshyponatriämie charakterisiert ist.
→ Ätiopathogenese:
→ I: Aufgrund unterschiedlicher Erkrankungen, aber auch Medikamenten-induziert kann es zur gesteigerten ADH-Sekretion kommen, die sekundär zu einer renalen Wasserretention führt. Gleichzeitig wird eine Verdünnungshyponatriämie durch Hemmung der Natriumrückresorption im proximalen Tubulus verstärkt. Grund hierfür ist eine Stimulation der Volumenrezeptoren bei Hypervolämie. Es kommt zur Ausscheidung eines inadäquaten konzentrierten Urins.
→ Klinisch-relevant: Das Schwartz-Bartter-Syndrom ist somit gekennzeichnet durch das Bestehen einer
→ A) Hyponatriämie bei Hypernatriurie und
→ B) Einer Plasmahypoosmolarität bei Hypervolämie.
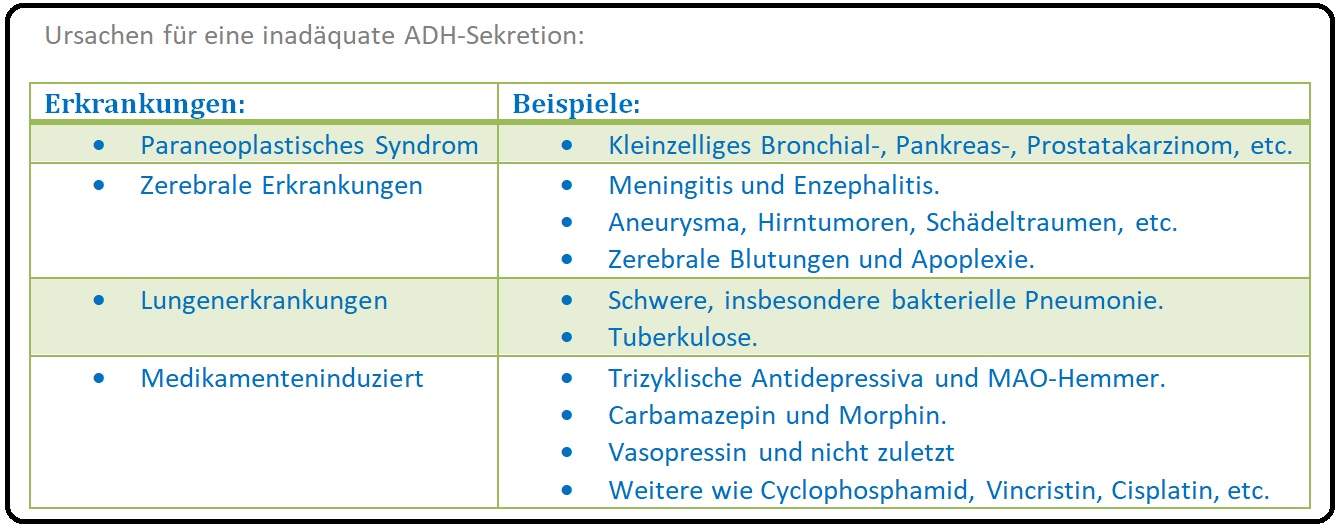
→ II: Ursachen für die Entwicklung eines SIADH sind u.a.:
→ 1) Paraneoplastische Syndrome: (stellt mit 80% der Fälle die häufigste Ursache dar). Durch Bildung von Arginin-Vasopressin und/oder vasopressinähnlichen Substanzen beim kleinzelligen Bronchialkarzinom (mit > 80% der Fälle die häufigste Ursache), Pankreaskarzinom, Prostatakarzinom, Blasenkarzinom etc.
→ 2) Zerebrale Erkrankungen: Durch Störungen des Zwischenhirn-Hypophysen-Systems mit konsekutiv gesteigerter ADH-Sekretion bei SHT, Subarachnoidalblutung, Sinusvenenthombose (v.a. Sinus-cavernosus-Thrombose), Meningitis (z.B eitrige Meningitis, tuberkulöse -, etc.), Enzephalitis, Hirnabszess, Guillain-Barre-Syndrom, Multiple-Sklerose, Delirium tremens, Hirnatrophie etc.
→ 3) Lungenerkrankungen: Pneumonie, TBC, Lungenabszess, Pneumothorax, zystische Fibrose.
→ 4) Medikamenteninduziert: Es ist auf den antidiuretischen Effekt der Medikamente zurückzuführen. Zu den Medikamenten gehören u.a. Vasopressin, Oxytocin, Thiazid-Diuretika, ACE-Hemmer, Cyclophosphamid, Vincristin, Morphin, Benzodiazepine, trizyklische Antidepressiva, Haloperidol, MAO-Hemmer, Carbamazepin etc.
→ 5) Weitere Erkrankungen: Sind Hypothyreose, Morbus Addison, schwere Hypophyseninsuffizienz, Myxödemkoma, etc.
→ Klinik: Die klinische Symptomatik ist Folge der hypotonen Hyperhydratation (= Wasserintoxikation) und abhängig von der Geschwindigkeit des Natriumabfalls.
→ I: Reduziert sich die Serumosmolalität langsam sind die Patienten meist lange beschwerdefrei und erst ab einer Na+-Konzentration < 120mmol/l treten Kopfschmerzen, Müdigkeit und Apathie auf.
→ II: Kommt es dagegen zu einem rapiden Abfall des Serumnatriums (< 120mmol/l) entwickeln sich charakteristische Symptome:
→ 1) Gastrointestinal: Appetitlosigkeit, Übelkeit, Erbrechen.
→ 2) Neurologisch: Kopfschmerzen, Muskelkrämpfe, Stupor, Somnolenz, Epilepsie und Koma.
→ 3) Psychiatrisch: Vermehrte Reizbarkeit, Persönlichkeitsveränderungen bis hin zu Verwirrtheitszustände, aber auch Depression.
→ Klinisch-relevant: Trotz des Vorliegens einer Hypervolämie bleibt die Entwicklung eines Aszites bzw. peripherer Ödeme aus. Des Weiteren ist der Urin trotz Flüssigkeitsüberladung konzentriert.

→ Diagnose:
→ I: Anamnese/Klinische Untersuchung: Sowohl die Eigenanamnese als auch der klinische Befund mit einer positiven Flüssigkeitsbilanzierung ohne periphere Ödeme lassen an ein SIADH denken. Des Weiteren ist die Medikamentenanamnese obligat. Bei der klinischen Untersuchung können insbesondere bei schwerer Hyponatriämie u.a. abgeschwächte Reflexe bis hin zu einem positiven Babinski-Reflex nachgewiesen werden.
→ II: Labor: Wegweisend ist die charakteristische Laborkonstellation bei normal funktionierender NNR und Niere bestehend aus:
→ 1) Serumhyponatriämie häufig < 130mmol/l,
→ 2) Eine deutlich reduzierte Blutosmolalität < 275mosmol/kg.
→ 3) Eine Bestimmung von ADH ist aufgrund der kurzen Halbwertszeit nicht zu empfehlen. Die ADH-Wirkung kann jedoch mit Hilfe der Harnosmolalität abgeschätzt werden; es zeigt sich eine erhöhter Urinosmolalität > 100mosmol/kg (Natriumkonzentration im Urin > 30mmol/l).
→ 4) Ggf. zusätzliche Bestimmung der Vasopressin- und Copeptin-Konzentration (= C-terminales Pro-Vasopressin-Fragment), die beim SIADH erhöht sind.
→ 5) ACTH-Kurztest zum Ausschluss eines Cortisol-Mangels.

→ Differenzialdiagnose: Vom Schwartz-Bratter-Syndrom müssen u.a. nachfolgende Erkrankungen abgegrenzt werden:
→ I: Renaler Salzverlust: Bei interstitieller Nephritis, renal-tubulärer Azidose, Hypoaldosteronismus, aber auch bei einer Therapie mit Diuretika, insbesondere Thiaziden.
→ II: Extrarenal: Bei Diarrhö, Erbrechen, Pankreatitis und Peritonitis.
→ III: Zerebral: Mit Natrium und Chlorid im Urin nach Kopfverletzungen und neurochirurgischen Eingriffen.
→ IV: Weitere Erkrankungen: Wie chronische Herzinsuffizienz, Leberzirrhose, nephrotisches Syndrom, aber auch Diabetes mellitus, psychogene Polydypsie, Hypothyreose, Mineralkortikoid-Mangel bei Morbus Addison, etc.
→ Klinisch-relevant: Beim nephrogenes Syndrom der inadäquaten Antidiurese (= NSIAD) handelt es sich um ein kongenital bedingtes Syndrom des Kindes und jungen Erwachsenen, das sich aufgrund einer Mutation des Arginin-Vasopressin-Rezeptor-2 entwickelt. Es besteht eine ähnliche klinische Symptomatik wie beim Schwartz-Bartter-Syndrom mit Hyponatriämie konzentriertem Urin, Kopfschmerzen, Apathie, Krampfanfällen, etc.; jedoch ist bei dieser Erkrankung das Plasma-ADH sehr niedrig, evtl. sogar nicht nachweisbar. Therapiemaßnahmen sind u.a. Flüssigkeitsrestriktion und die Substitution von Harnstoff zur osmotischen Diurese.
→ Therapie:
→ I: Primär steht bei der Behandlung des SIADH die Beseitigung der Ursache im Vordergrund. Wenn möglich, sollten die auslösenden Medikamente abgesetzt werden.
→ II: Symptomatische Therapie:
→ 1) Ziel ist es, die negative Wasserbilanzierung und die damit verbundene Hyperhydratation durch Beschränkung der Wasserzufuhr (800-1000ml) aufzuheben. Folge ist ein Anstieg der Na+-Serumkonzentration und Osmolalität, die konsekutiv zu einer Besserung der klinischen Symptomatik führt.
→ 2) Eine weitere Therapieoption ist die Applikation eines Vasopressinrezeptor-Anatagonisten (= Vaptane z.B. Tolvaptan), der am Vasopressin-Rezeptor die ADH-Wirkung hemmt und die Zunahme der Aquarese und den Anstieg der Natrium-Serumkonzentration induziert.
→ III: Schwere Hyponatriämie:
→ 1) Besteht eine ausgeprägte Hyponatriämie (Na+-Konzentration < 100mmol/l) mit charakteristischer klinischer Symptomatik (z.B. Verwirrtheitszustände, Epilepsie, Koma) ist die vorsichtige Substitution von 150ml einer 3%igen, hypertonen Na+Cl--Lösung indiziert.
→ Klinisch-relevant: Hierbei darf der Natrium-Serumspiegel nur um maximal 10mmol/l in 24 Stunden angehoben werden, da sich ansonsten eine zentrale pontine Myelinolyse ausbilden kann.
→ 2) Gegebenenfalls kann zusätzlich Furosemid zur Steigerung der Diurese verabreicht werden.
- Details
- Kategorie: Hypophyse
- Zugriffe: 8436
→ Definition: Die Hyperprolaktinämie beschreibt eine Erhöhung der Prolaktin-Konzentration über den Normbereich von 2-25µg/l (pathologische Prolaktin-Konzentration > 200µg/l). Beim Prolaktinom handelt es sich (überwiegend) um ein Prolaktin-produzierendes Adenom des Hypophysenvorderlappens mit konsekutiver Hyperprolaktinämie.
→ Epidemiologie:
→ I: Das Prolaktinom stellt den häufigsten endokrin-aktiven Hypophysentumor (Adenom) dar und ist in 20% der Fälle Ursache einer sekundären Amenorrhoe.
→ II: Die Indizidenz liegt in Deutschland bei 5-6/100000 Einwophner pro Jahr.
→ II: Frauen (überwiegend Mikroprolaktinome mit geringem Wachstum) sind deutlich häufiger betroffen als Männer (überwiegend Makroadenome), wobei der Manifestationsgipfel zwischen dem 20.-40. Lebensjahr liegt.
→ Ätiopathogenese: Prolaktin-produzierendes Hypophysenadenom:
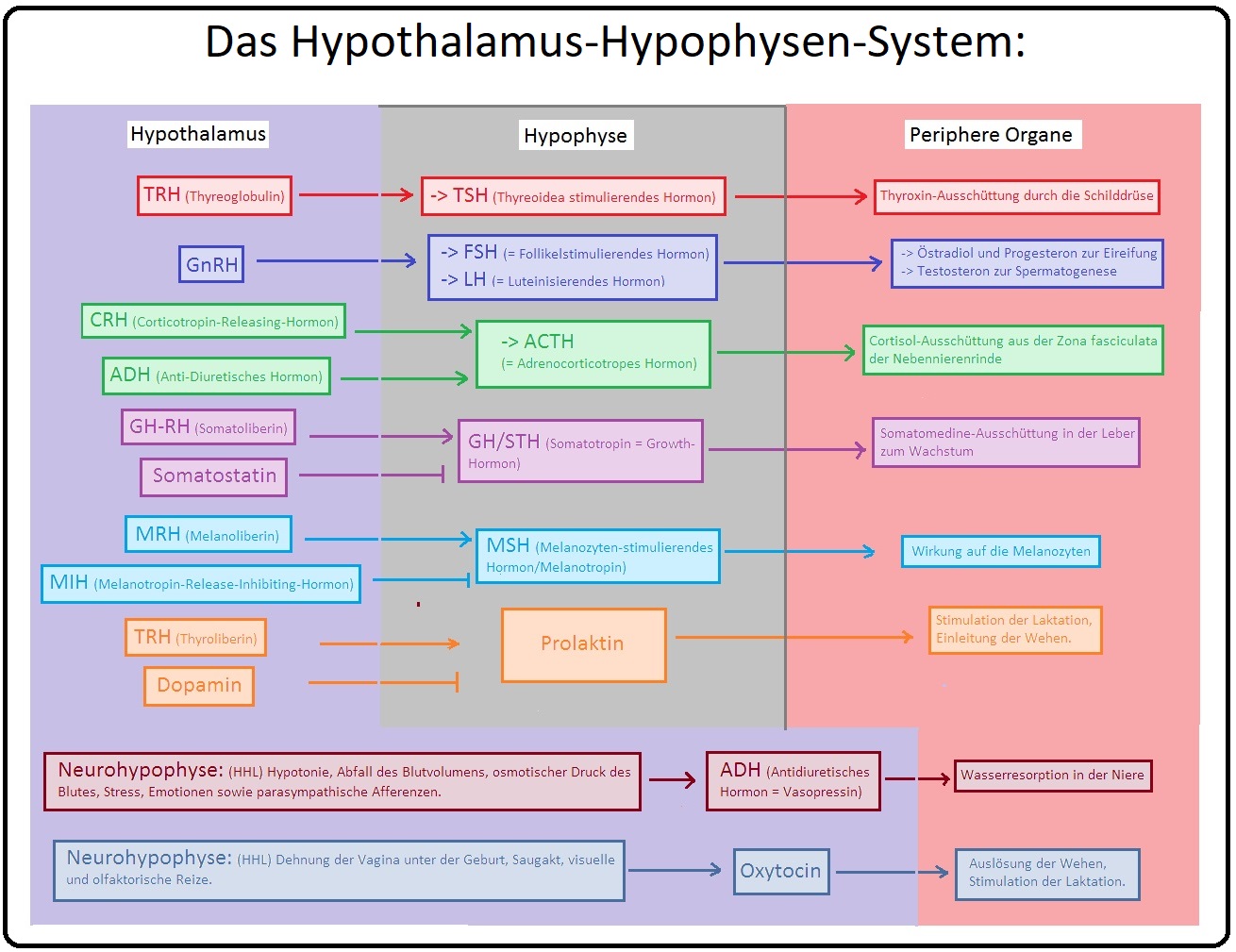
→ I: Prolaktin leitet die Laktation der Brustdrüse ein und wird durch den Hypothalamus reguliert. Regulatorische Mechanismen sind u.a.:
→ 1) Prolactin-inhibiting-factor (= Dopamin) hemmt die Ausschüttung,
→ 2) TRH fördert die Ausschüttung von Prolaktin.
→ II: Häufigste physiologische Ursache für eine Hyperprolaktinämie ist die Schwangerschaft mit Anstieg des Prolaktinspiegels auf das 10-fache des Normwertes. Auch der Saugreiz beim Stillen ruft eine Hyperprolaktinämie hervor.
→ III: Beim Prolaktinom entwickelt sich über eine hypothalamisch-unabhängige (= autonome) Prolaktinproduktion und -sekretion eine Beeinträchtigung der Hypophysen-Gonaden-Achse. Dies wiederum führt zur:
→ 1) Hemmung des Zyklus-bedingten LH-Anstiegs mit Anovulation und Hypoöstrogenämie.
→ 2) Ausbildung eines Hypogonadismus.
→ IV: Weitere Ursachen:
→ 1) Entzügelungshyperprolakinämie: Hierbei kommt es zur Kompression des Hypopyhsenstiels aufgrund von para- oder suprasellären Tumoren, die zu einer Unterbindung der Dopamin-Zufuhr, das als Prolactin-inhibiting-factor agiert, führen.
→ 2) Chronische Niereninsuffizienz mit konsekutiv verminderten Prolaktinclearance.
→ 3) Funktionelle Hyperprolaktinämie bei fortgeschrittener Leberzirrhose.
→ 4) Primäre Hypothyreose führen über einen erhöhten TRH-Spiegel zu gesteigerten Prolaktinspiegeln.
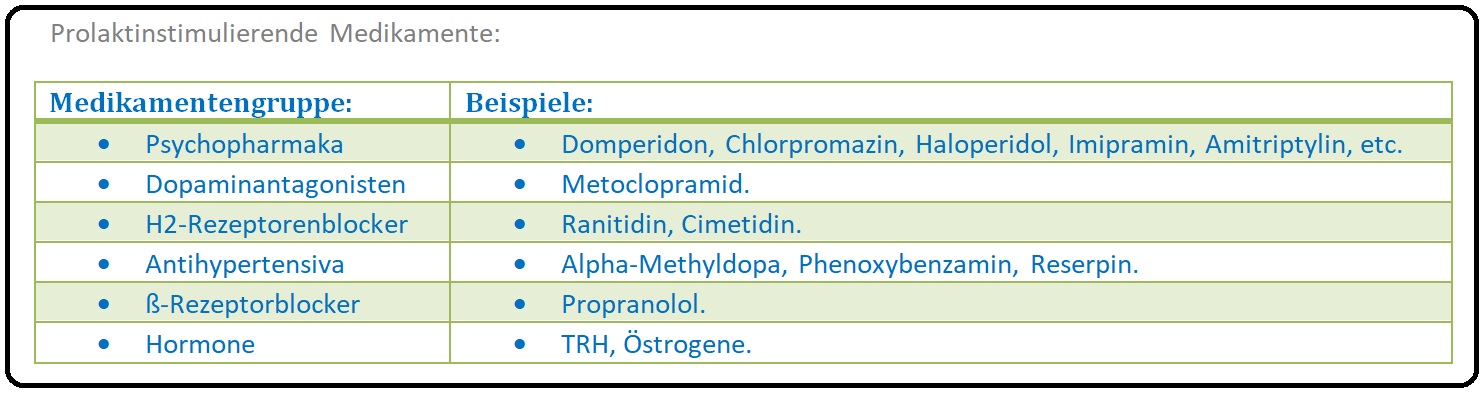
→ 5) Medikamenten-induziert durch dopaminantagonistische Substanzen wie Antidepressiva, Neuroleptika, (z.B. Haloperidol), die mit Dopamin um den Rezeptor konkurrieren, aber auch atypische Neuroleptika, Antiemetika, Opiate, Kalziumantagonisten, etc.
→ Klassifikation:
→ I: Mikroprolaktinom:
→ 1) Der Durchmesser ist < 1cm;
→ 2) Sie bleiben häufig größenkonstant mit einem stabilen Prolaktinspiegel.
→ 3) Stellt mit 70% die häufiger Form dar und treten zumeist beim weiblichen Geschlecht auf.
→ II: Makroprolaktinom:
→ 1) Der Durchmesser ist > 1cm; die Prolaktin-Konzentration liegt meist > 200ng/ml und es manifestiert sich eine anhaltende Proliferation.
→ 2) Makroprolaktinome treten bei Frauen und Männern gleich häufig auf.
→ 3) Hierbei stehen insbesondere die Folgen (abhängig von der Tumorgröße) der Raumforderung mit z.B. Kopfschmerzen, Hirnnervenausfällen, Blickparesen, Gesichtsfeldausfällen, etc im Vordergrund)
→ Klinik: Leitsymptom des Prolaktinoms ist der Hypogonadismus mit einer charakteristischen klinischen Symptomatik:
→ I: Frauen: Klassische Symptome sind Zyklusstörungen mit Anovulation, sekundärer Oligo- bis Amenorrhoe, Infertilität, Galaktorrhoe und Libidoverlust, aber auch Hirsutismus und Akne.
→ II: Männer: Störungen der Libido und Potenz bis hin zur Impotenz, sowie eine Gynäkomastie.
→ III: Spätsymptome: Als lokale Komplikation der Raumforderung sind u.a. chronische Kopfschmerzen, evtl. Störungen des Gesichtsfeldes (bilaterale Hemianopsie) sowie Ausfälle weiterer Hirnnerven, insbesondere die Augenmuskelparese. Weitere Spätsymptome sind Osteoporose bei langjährigem Hypogonadismus, psychische Störungen mit ängstlichen Verstimmungszuständen und Depression, aber auch Zeichen einer Hypophyseninsuffizienz mit Verminderung oder Ausfall der Sekretion von GH, ACTH, FSH, LH (= Hypopituitarismus).
→ Diagnose:
→ I: Anamnese/klinische Untersuchung: Medikamentenanamnese (z.B. Östrogene, Neuroleptika, Metoclopamid, Alpha-Methyldopa, etc.), Ausschluss einer primären Hypothyreose, sowie einer chronischen Niereninsuffizienz bzw. Leberzirrhose.
→ II: Labor:
→ 1) Mehrfache Bestimmung der Prolaktinkonzentration. Werte > 200ng/ml sind beweisend; Werte zwischen 25-200ng/ml bedürfen einer weiteren Abklärung.

→ Klinisch-relevant:
→ A) Prolaktin unterliegt einer zirkadianen Rhythmik; während des Schlafes, vor allem früh morgens ist die Konzentration am höchsten und nimmt über den Tag hin ab. Eine Prolaktinbestimmung sollte aufgrund dessen erst 1-2 Stunden nach dem Aufwachen erfolgen.
→ B) Stressoren wie starke Schmerzen, etc., aber auch eine Hypothyreose können zu einem Prolaktinanstieg führen.
→ 2) TRH-Test: Zur Kontrolle der laktotropen Achse (bzw. der hypophysären Funktion). Bestimmung des Prolaktin-Basal-Wertes; anschließend Gabe von 200µg TRH intravenös. und nach 30min. erneute Blutabnahme zur Prolaktinbestimmung. Typischerweise bleibt beim Prolaktinom der Prolaktin-Anstieg aus.
→ 3) Weitere Laborparameter sind TSH, Kreatinin und ein Schwangerschaftstest.
→ III: CT/MRT: Zur Tumorsuche; ist die Untersuchung positiv sollte eine Kontrolle der Hypophysenpartialfunktionen durch Bestimmung der basalen Hormonkonzentrationen erfolgen.
→ Differenzialdiagnose: Weitere Ursachen, die zu einer Hyperprolaktinämie führen können:
→ I: Hyperprolaktinämie: Aufgrund einer verminderten hypothalamischen Hemmung; para- und supraselläre Tumoren können durch Kompression bzw. Zerstörung der Prolactin-inhibiting-factor (= Dopamin) produzierenden Zellen eine Hyperprolaktinämie induzieren.
→ II: Medikamentös-induziert: Durch Neuroleptika (gerade Haloperidol), Antidepressiva (Imipramin, Amitriptylin), Antiphypertensiva wie Alpha-Methyldopa, Reserpin, Verapamil, ß-Blocker (Propranolol), Antiemetika (Metoclopramid, Domperidon), Antihistaminika (Ranitidin, Cimetidin) oder Östrogen induziert.
→ III: Funktionell: Im Stadium der chronischen Niereninsuffizienz und fortgeschrittenen Leberzirrhose,
aber auch im Zusammenhang einer MEN-I (= Wermer-Syndrom).
→ IV: Bei der primären Hypothyreose (eine primäre Hypothyreose führt zu einem Anstieg des TRH, welches eine positive Wirkung auf die Prolaktinsekretion hat).
→ V: Bei Galaktorrhoe muss auch immer ein Mamma-Karzinom ausgeschlossen werden.
→ VI: Physiologisch: In der Schwangerschaft und Stillzeit.
→ Therapie:
→ I: Medikamentös: Die Behandlung mit einem Dopamin-Antagonisten führt nicht nur zu einer Prolaktinkonzentrationsreduktion, sondern hat auch einen antiproliferativen Effekt mit Größenabnahme des Tumors.
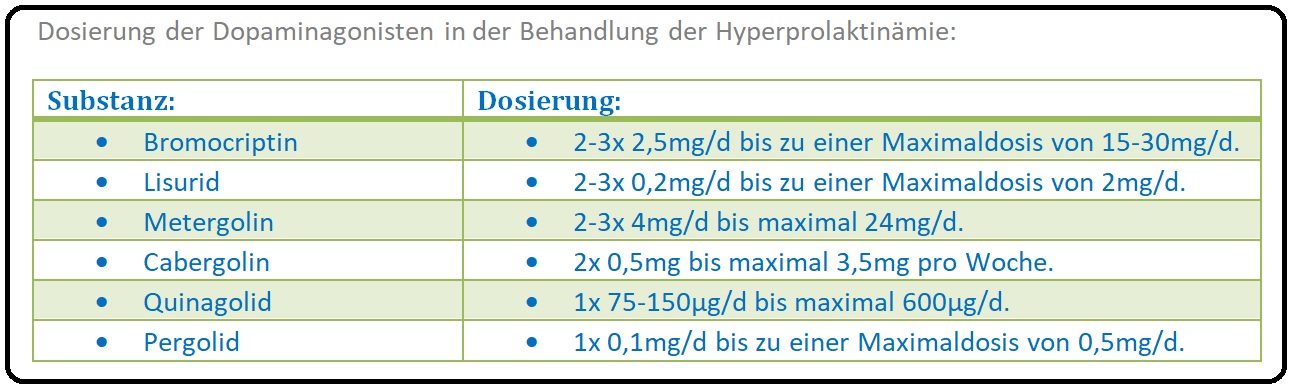
→ 1) Als Mittel der 1.Wahl haben sich Dopamin2-Rezeptoragonisten wie Bromocriptin, Quinagolid oder Cabergolin etabliert.
→ 2) Sie führen frühzeitig zu einer Senkung des Prolaktinspiegels sowie zu einer Reduktion der Tumorgröße.
→ 3) Dosierung: Es erfolgt eine einschleichende Therapie, um mögliche Nebenwirkungen wie (Übelkeit, Erbrechen, orthostatische Dysregulation) zu vermeiden.
→ 4) Nach 2-3 Jahren kann bei normaler Prolaktinkonzentration ein Auslassversuch (kontrovers diskutiert) angestrebt werden. Es sind jedoch engmaschige Prolaktin-Kontrollen obligat. Zumeist wird die pharmakologische Behandlung lebenslang fortgesetzt.
→ II: Operativ: Eine transsphenoidale Adenomektomie ist nur bei Unverträglichkeit oder erfolgloser medikamentöser Therapie (bestehende Gesichtsfeldausfälle) indiziert.
→ III: Bestrahlung: Die konventionelle externe Bestrahlung erfolgt nur bei invasiv wachsenden Makroadenomen, die durch eine medikamentöse oder chrirurgische Therapie in Bezug auf das Tumorwachstum und die Prolaktinsekretion nicht beherrscht werden können.
→ Prognose: Gerade bei den Mikroadenomen kann durch eine adäquate medikamentöse Therapie eine Normalisierung des Menstruationszyklus und der Ovulation erreicht werden.
- Details
- Geschrieben von: CF
- Kategorie: Hypophyse
- Zugriffe: 6385
→ Definition: Bei der Akromegalie handelt es sich um ein unproportionales Wachstum der Akren nach Abschluss des physiologischen Längenwachstums zumeist aufgrund eines GH-(Somatotropin)-produzierenden Hypophysenadenom. Tritt diese GH-Überproduktion vor Beendigung des Wachstums (vor dem Schluss der Epiphysenfungen) auf wie bei Kindern, spricht man von hypophysärem Gigantismus mit einer Körpergröße > 2m.
→ Epidemiologie:
→ I: Wachstumsproduzierende Hypophysenadenome sind die 2.-häufigsten endokrin aktiven Hypophysentumoren.
→ II: Die Inzidenz liegt bei 0,3/100000 Einwohnern pro Jahr und stellt somit eine sehr seltene Erkrankung dar. In 5% der Fälle kiegt eine Assoziation mit MEN-1 vor.
→ III: Die Akromegalie kann in jedem Alter auftreten, zeigt jedoch einen Manifestationsgipfel zwischen der 3.-4. Lebensdekade (wobei Frauen wie Männer sind gleichermaßen betroffen sind).
→ Physiologie:
→ I: Das Hormon GH ist ein aus 191 AS bestehendes Polypeptid und wird insbesondere im Schlaf sezerniert, während am Tag die Konzentration am niedrigsten ist. Des Weiteren führen Hunger, körperlicher Anstrengung und Stress, etc. zu einer vermehrten GH-Ausschüttung (= Growth-Hormon); durch Nahrungsaufnahme (Hyperglykämie) wiederum wird die GH Sekretion gehemmt.
→ II: Die Sekretion von GH wird durch das GH-Releasing-Hormon (= GHRH) stimuliert und durch Somatostatin gedrosselt.
→ III: Die Wirkung von GH wird zum einen indirekt durch den Insulin-like-growth-Faktor 1 (= IGF-1) hervorgerufen, zum anderen beeinflusst GH direkt einige Stoffwechselvorgänge wie den Einbau von Aminosäuren in Proteine oder Freisetzung freier Fettsäuren aus den Fettzellen. IGF-1 wird fast ausschließlich in der Leber produziert wird und induziert die Blockade der GH-Sekretion im Sinne einer negativen Rückkopplung.
→ Ätiopathogenese: Ursache dieses Wachstumshormonexzesses sind zumeist Hypophysenadenome (in der Regel mit 95% der Fälle benigne Somatotropin-produzierende Tumoren des Hypophysenvorderlappens) oder sehr seltene extrahypophysäre Tumore (hypothalamisch, paraneoplastisch insbesondere bei Bronchial-, Mamma- und Pankreaskarzinom), die GHRH produzieren. Folge ist eine massiv gesteigerte Bildung und Freisetzung von IGF-1, die konsekutiv zum pathologischen Wachstum von Bindegewebe, Haut, Skelett und inneren Organen führen.

→ Klinik: Die klinische Symptomatik der Akromegalie lässt aus der Wirkung von GH bzw. seines Zielhormons IGF-1 (überwiegend hepatisch gebildet) ableiten. Nicht selten dauer die Diagnosestellung 5-10 Jahre, da sich die klinische Symptomatik nur schleichend entwickelt und vom Paitenten selbst kaum wahrgenommen wird.
→ I: Charakteristische Leitsymptome sind u.a. verdickte, faltige Gesichtshaut (= Cutis gyrata), Vergröberung der Gesichtszüge insbesondere der Nasolabialfalte, Vergrößerung von Händen, Füßen und des Schädels aufgrund eines appositionellen Knochenwachstums sowie der Zunge (= Makroglossie) mit kloßiger Sprache. Des weiteren manifestiert sich im weiteren Krankheitsverlauf eine Größenzunahme der inneren Organe wie die Viszeromegalie und Kardiomegalie (z.B. Kardiomyopathie).
→ II: Fakultative Symptome: Sind vor allem:
→ 1) Arterielle Hypertonie, Kopfschmerzen, Sehstörungen, Gesichtfeldausfälle (= bitemporale Hemianopsie durch Kompression des Chiasmus opticum) sowie Hirnnerv- und nicht zuletzt Müdigkeit durch ein zentrales Schlafapnoe-Syndrom.
→ 2) Knochenschmerzen, Chondrokalzinose sowie das Karpaltunnelsyndrom mit Parästhesien durch Kompression des N. medianus.
→ 3) Hyperhidrosis und Hypertrichosis.
→ 4) Evtl. pathologische Glukosetoleranz mit Tendenz zur Entwicklung eines Diabetes mellitus Typ II sowie
→ 5) Evtl. Auftreten einer sekundären Amenorrhoe, Symptomen eines Hypogonadismus sowie Potenz- und Libidostörungen durch Zerstörung der gonadotropinbildender Hypophysenzellen.
→ 6) Auch kann sich einer Hyperprolaktinämie (in bis zu 75%) durch Kompression des Hypophysenstiels entwickeln.
→ 7) Proximale Myopathie, generalisierte Muskelschwäche und vorzeitige Ermüdbarkeit (die Enwicklung der Myopathie ist insbesondere abhängig von der Dauer der Akromegalie) treten in einem späten Stadium der Akromagelie auf.
→ III: Komplikation: Eine wichtige und z.T. schwerwiegende Komplikation ist die Entwicklung einer Kardiomyopathie.

→ Diagnose:
→ I: Anamnese und klinische Untersuchung.
→ II: Labor:
→ 1) IGF-1 Bestimmung (Serum tiefgefroren); Serumkonzentrationen < 5ng/ml machen eine Akromegalie unwahrscheinlich; des Weiteren kann das GH-abhängige Bindungsprotein 3 (IGFBP-3) mitbestimmt werden.
→ 2) Serum GH erhöht aufgrund der schwankenden Hormonausschüttung müssen mehrere Werte im Tagesprofil genommen werden.
→ 3) GH-Bestimmung: Unter Glukosebelastung (oraler Glukosetoleranztest). Sie erfolgt nach einer Nahrungskarenz von 12 Stunden und basaler GH-Bestimmung und anschließender Gabe von 75g Glukose. Im Anschluss wird nach 30, 60 und 90min GH bestimmt. Eine Akromegalie ist wahrscheinlich, wenn GH nicht unter 2ng/ml fällt. Bei Suppression < 1ng/ml ist eine autonome GH Sekretion fast ausgeschlossen.
→ 4) Zum Ausschluss von Unterfunktionszuständen müssen ebenfalls TSH, fT4, Kortisol, LH, FSH, Testosteron mitbestimmt werden.
→ 5) Nachweis einer Hyperphosphatämie (angenommen wird eine erhöhte renale Phosphatresorption duch IGF-1).
→ III: Bildgebende Verfahren: CT und MRT gelten als Goldstandard in der Diagnostik dieses Hypophysentumors (bezüglich der Größe, Lage und Ausdehnung). Die Sella ist in 90% der Fällen vergrößert.
→ IV: Pathologie: Mikroskopisch kann zwischen wenig granulierten (aggressiv) und einem dicht granulierten (weniger aggressiv) Tumoren differenzierten werden.
→ V: Augenärztliche Untersuchung zur Diagnose eines möglichen Gesichtsfelddefektes.
→ Differenzialdiagnose: Die Akromegalie muss insbesondere von nachfolgenden Erkrankungen abgegrenzt werden; hierzu zählen:
→ I: GH-Überproduktion ohne Adenome bzw. bei Hypophysenhyperplasie
→ II: Ektope oder paraneoplastische GH- bzw. GHRH-Produktion. GH-Erhöhungen findet man unter anderem auch bei:
→ 1) Angst und körperlicher Belastung physiologischerweise.
→ 2) Doping mit Wachstumshormonen.
→ 3) Erkrankungen insbesondere chronischer Niereninsuffizienz und Leberzirrhose.
→ III: Akromegalie im Rahmen eines MEN-1-Syndroms.
→ IV: Es muss aber auch die Abgrenzung zur Dystrophia deformans Paget erfolgen.
→ Therapie: Das Behandlungsziel der Akromegalie ist die Normalisierung der IGF-1- und GH-Konzentration. Zudem sollten Komorbiditäten diagnostiziert und therapiert werden, um sowohl die Lebensqualität, als auch die Mortalität zu verbessern.
→ I: Mittel der Wahl ist die operative transsphenoidale Adenomektomie (kurative Therapie). Wichtige Komplikationen bei der operativen Therapie sind Liquorfistel, Hypophysenvorderlappeninsuffizienz, Meningitis und persistierender Diabetes insipidus. Bei Inoperabilität oder unzureichendem Operationserfolg kann eine Strahlentherapie indiziert sein.
→ II: Strahlentherapie: Bei Therapieversagen oder erneutem Tumorwachstum wird eine Strahlentherapie empfohlen. Gängige Verfahren hierbei sind die konventionelle, fraktionierte Röntgenbestrahlung oder die Radiochirurgie (z.B. Linearbeschleuniger oder "Gamma-Knife"). Durch den deutlich verzögerten Wirkeintritt nach Monaten bis Jahren muss initial zusätzlich medikamentös therapiert werden. Mögliche Nebenwirkungen der Strahlentherapie sind insbesondere Insuffizienz der Hypophysenfunktion, Optikusneuropathie und Erblindung, etc.
→ III: Medikamentöse Therapie: Die medikamentöse Behandlung ist indiziert bei z.B. unzureichendem OP-Erfolg, Kontraindikationen, Inoperabilität oder als überbrückende Maßnahme nach Strahlentherapie. Ziel hierbei ist die Hemmung der GH-Sekretion bzw. -wirkung.
→ 1) Dopamin-2-Agonisten: Wie Bromocriptin sind nur in 20% der Fälle erfolgreich, da häufig eine ausreichende GH-Senkung durch Hemmung nicht erreicht wird. Sie werden überwiegend zur Kombination mit Somatostatinanaloga verabreicht, wenn diese nicht ausreichend wirken.
→ 2) Somatostatin-Analoga: (hierzu zählen Octreotid, Lanreotid, etc). Octreotid hemmt die GH-Sekretion kanninitial 2-3x täglich subkutan appliziert werden, und anschließend als Depotpräparat 1x monatlich intramuskulär. Verwendung finden die Somatostatinanaloga insbesondere zur präoperativen Vorbereitung, zur Überbrückung bis zum Wirkeintritt einer Strahlentherapie sowie bei Kontraindikationen, etc.
→ 3) GH-Rezeptor-Analoga: Pegvisomant (tägliche s.c. Injektion) hemmt die GH-Wirkung am Rezeptor und führt konsekutiv zu einer Normalisierung des IGF-1-Spiegels (vermindert jedoch nicht die GH-Sekretion); Folge ist eine deutliche klinischen Besserung. Sie reduzieren aber nicht die Größe des Hypophysenadenoms.
→ Prognose: Unbehandelt verkürzt die Akromegalie die Lebenserwartung um ca. 10 Jahre insbesondere aufgrund von kardialen und zerebrovaskulären Komplikationen (z.B. arterielle Hypertonie, Kardiomegalie, Schlafapnoesyndrom, etc.). Auch zeigt sich ein vermehrtes Auftreten von Mamma- und Kolonkarzinomen. Die Normalisierung des Wachstumshormons IGF-1 verbessert die Prognose.
- Details
- Kategorie: Hypophyse
- Zugriffe: 15059
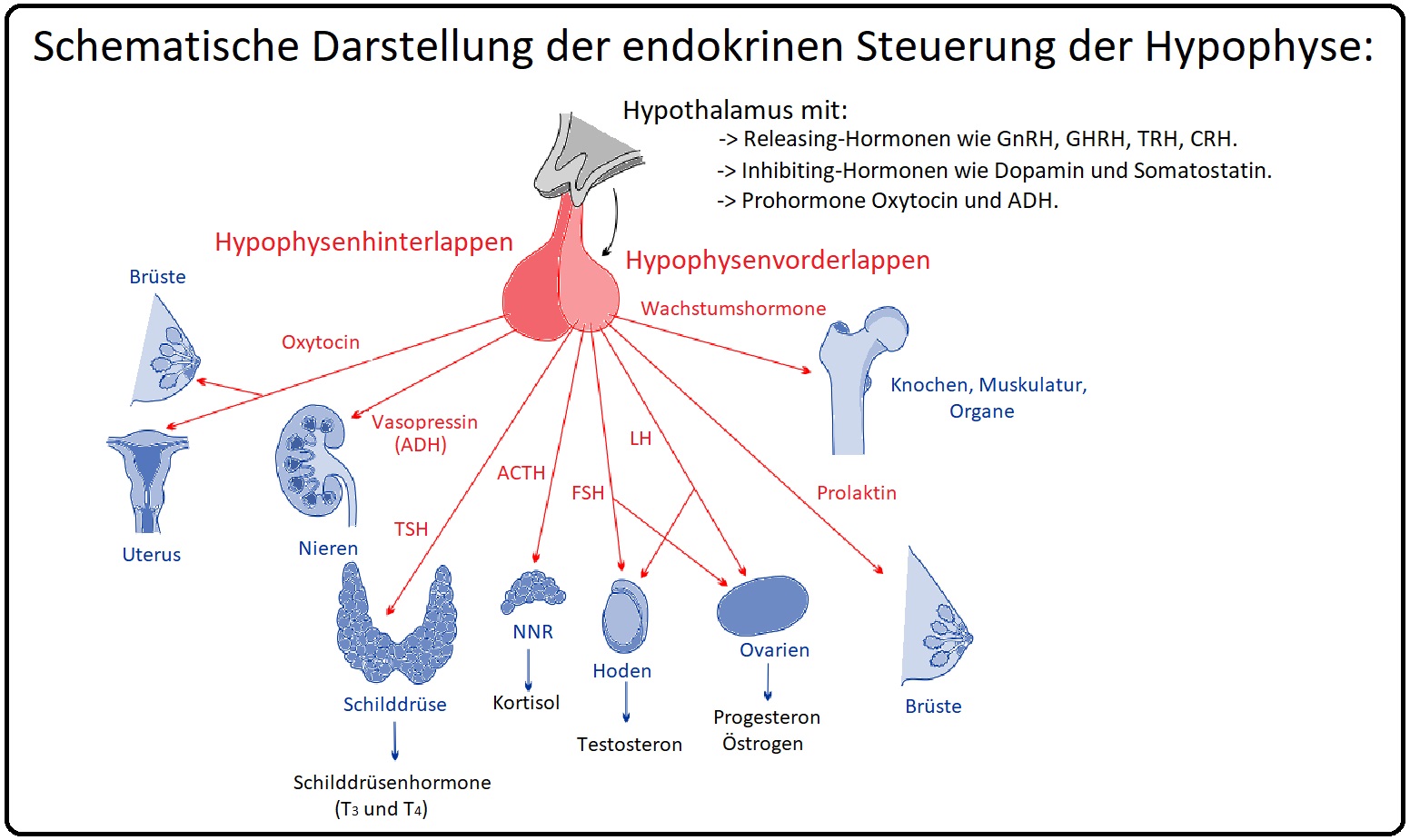
→ Definition: Bei der HVL-Insuffizienz handelt es sich um einen partiellen oder totalen Funktionsausfall der Hypophyse infolge einer verminderten bzw. fehlenden Sekretion adenotroper Hormone. Das Sheehan-Syndrom wiederum stellt eine postpartale Hypophysennekrose dar. Während der Schwangerschaft wächst die Hypophyse und reagiert empfindlich auf Hypoxie (Ursache hierfür sind insbesondere größerer Blutverlust und Thrombosebildungen).
→ Ätiologie: Ursachen für die Entstehung eines Hypopituitarismus sind u.a.:
→ I: Tumorös: Raumforderungen wie Hypophysenadenome, Kraniopharyngeome (benigner Tumor, der sich aus Restzellen der Rathke-Tasche entwickelt), Meningeome oder Metastasen.
→ II: Traumatisch: Traumen (z.B. Schädel-Hirn-Trauma) im Bereich der Hypophyse mit Einblutungen, Operationen, Bestrahlung.
→ III: Vaskulär: Wichtig zu erwähnen, ist das Sheehan-Syndrom, welches nur noch selten auftritt. Hierbei entwickelt sich eine Hypophysenvorderlappeninsuffizienz infolge eines ausgeprägten postpartalen Blutverlustes mit anschließender Nekrosebildung. Aber auch beim A. carotis Aneurysma und der Arteriitis temporalis (Horton)
→ IV: Entzündlich: Autoimmunhypophysitis (tritt charakteristischweise in der 2. Schwangerschaftshälfte auf mit lymphozytären Infiltrationen), granulomatöse Systemerkrankungen wie Sarkoidose, Wegener-Granulomatose und TBC.
→ V) Weitere Ursachen: Sind u.a. Amyloidose, Hämochromatose oder post chirurgisch.
→ Klassifikation:
→ I: Nach der Entstehung:
→ 1) Primäre HVL-Insuffizienz: Hierbei sind die Zellen der Hypophyse selbst betroffen.
→ 2) Sekundäre HVL-Insuffizienz: Durch Störungen bzw. Kompression des Hypothalamus oder des Hypophysenstiels.
→ II: Nach ihrem Funktionsausfall:
→ 1) Partielle HVL-Insuffizienz: Mit Störung der Sekretion einer oder mehrerer HVL-Hormone.
→ 2) Komplette HVL-Insuffizienz: Mit Beeinträchtigung aller Hypophysenvorderlappen-Hormone (GH, LH, FSH, TSH, ACTH, PRL); wird als Panhypopituitarismus bezeichnet.
→ Klinisch-relevant: Klinische Symptome manfestieren sich erst, wenn 80% des Hypophysenvorderlappens zerstört sind. Besteht der Übergang von einer partiellen in eine komplette HVL-Insuffizienz fallen die Hormone in folgender Reihenfolge aus; beginnend mit GH und Gonadotropin (FSH, LH) bis schließlich zum ACTH. Prolaktin fällt nur selten aus.
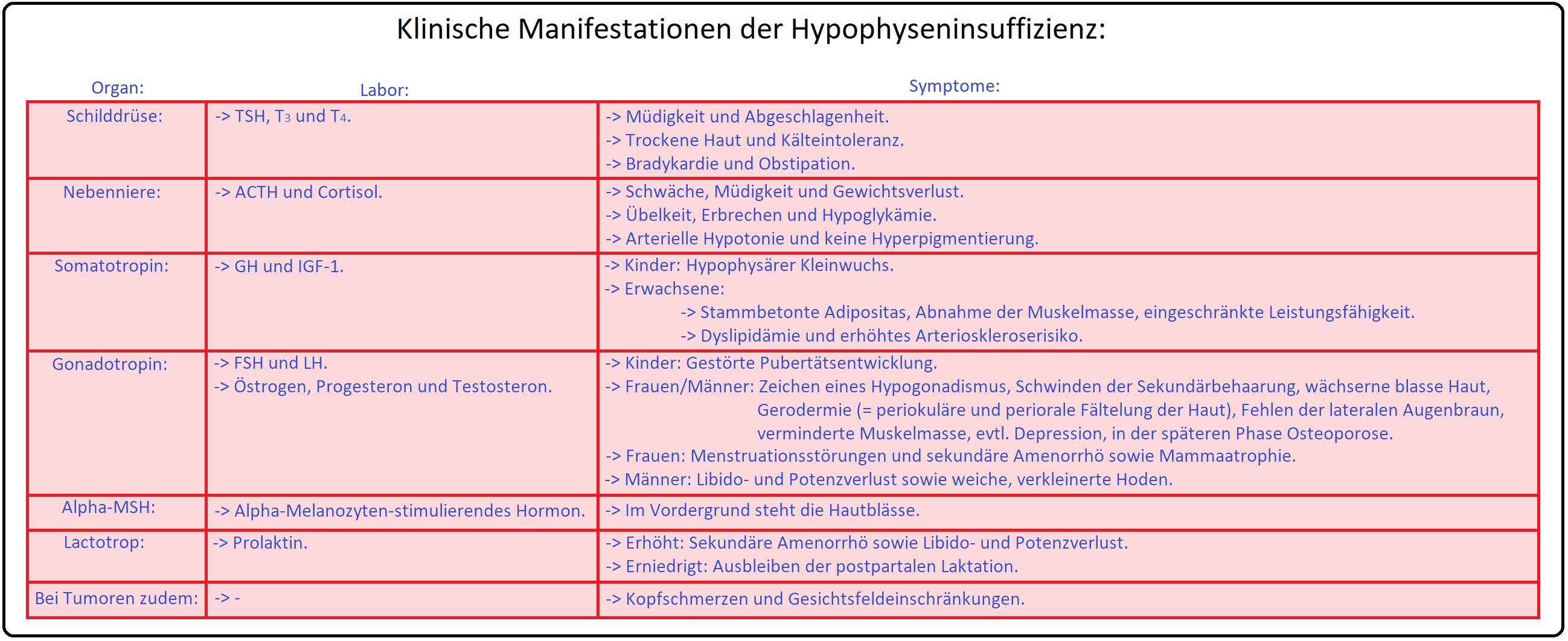
→ Klinik: Die klinische Symptomatik ist vor allem abhängig vom Ausfall des Hormons sowie vom Zeitpunkt (Alter: Präpubertät oder Postpubertät); Klinische Symptome treten erst und zumeist schleichend auf, wenn 80% des Hypophysenvorderlappens zerstört ist. Bei den Hypophysenadenomen manifestiert sich eine charakteristische Reihenfolge des Funktionsausfalls, beginnend mit GH über FSH bzw. LH und TSH zu ACTH.
→ I: Partieller Ausfall:
→ 1) STH-Ausfall: Manifestiert sich vor der Pubertät eine STH-Sekretionsstörung, entwickelt sich ein hypophysären Minderwuchs bei normalen Körperproportionen und Intelligenz. Im Erwachsenenalter bilden sich wie abdominelle Fetteinlagerung, erhöhtes Arteriosklerose-Risiko (LDL erhöht, HDL-erniedrigt) aus.
→ 2) TSH-Ausfall: Ausbildung einer sekundäre Hypothyreose mit u.a. trockener teigiger Haut, Kältemepfindlichkeit, psychomotorischer Verlangsamung und periorbitalem Ödme.
→ 3) ACTH-Ausfall: Manifestation einer sekundären Nebenniereninsuffizienz und konsekutiven klinischen Zeichen wie frühzeitige Ermüdbarkeit, Adynamie, Gewichtsverlust, Pigmentstörungen (wächserne Blässe) Hypotonie und Hypoglykämie.
→ 4) Gonadotropin-Ausfall: Präpubertär manifestiert sich eine ausbleibende Geschlechtsreife (= Eunuchoidismus = Kallman-Syndrom). Postpubertär sind klassisch klinische Symptome Zyklusstörungen, sekundäre Amenorrhö, Libido- und Potenzstörungen (= Hypogonadismus) sowie der Verlust der Achsel- und lateralen Augenbraunbehaarung.
→ Klinisch-relevant: Frühsymptome des Sheehan-Syndroms sind Agalaktie, Amenorrhö und Verlust der sekundäre Behaarung. Die Symptome können sich z.T. erst nach Jahren manifestieren.
→ II: Kompletter Ausfall: Hierbei sind charakteristische klinische Symptome des Hypopituitarismus Müdigkeit, Abgeschlagenheit, trockene kühle dünne Haut. Aufgrund des ACTH-Mangels zeigt sich eine alablasterfarbene Haut, Verlust der sekundären Schambehaarung, evtl. Ausbleiben der Bartbehaarung sowie das Auftreten einer Hypotonie und Hypoglykämie.
→ Klinisch-relevant: Beim Hypopituitarusmus treten die 7 As auf:
→ A) Gonadotropin-bedingt: Verlust der Achsel- und lateralen Augenbraunbehaarung (= Herthoge-Zeiche), Amenorrhö, Agalaktie.
→ B) TSH-bedingt: Apathie,
→ C) MSH-bedingt: Alablastärfarbene, blasse Haut.
→ D) ACTH-bedingt: Adynamie.
→ III: Weitere Symptome: Im Rahmen von Raumforderungen manifestieren sich im weiteren Krankheitsverlauf zudem ausgeprägte Kopfschmerzen und Gesichtsfeldausfälle, etc.
→ IV: Komplikation: Wichtigste und schwerwiegenste Komplikation ist das unter Stresssituationen (z.B. Infektionen, großer Operationen, etc.) sich entwickelnde hypophysäre Koma (durch Dekompensation einer chronischen HVL-Insuffizienz).
→ Diagnose: Bei Verdacht auf eine Hypophyseninsuffizienz bzw. einen Hormonmangel sollten diagnostisch (immer) sowohl die hypophysären als auch die peripheren Hormone (gleichzeitg) bestimmt werden.
→ I: Anamnese und klinische Untersuchung.
→ II: Labor: Mit insbesondere:
→ 1) Verminderung der peripheren Hormonkonzentrationen wie fT3, fT4, Cortisol, Östradiol und Testosteron.
→ 2) Erniedrigung der basalen Hypophysenhormonspiegel für FSH, LH, TSH und ACTH.
→ III: Endokrine-Stimulationstests: Bei Makroadenomen ist zu berücksichtigen, dass es durch einen Stimulationstest potenziell zu einem Hypophysenapoplex kommen kann.
→ 1) Somatotrope Funktion: Nach Applikation von GHRH oder im Insulin-Hypoglykämie-Test sind ein unzureichender Anstieg von IGF-1 und GH eruierbar.
→ 2) Gonadotrope Funktion: LH und FSH basal sowie Östradiol und Testosteron sind nach Gabe von LHRH erniedrigt.
→ 3) Thyreotrope Funktion: Nach Applikation von THR kommt es zum unzureichenden Anstieg von TSH sowie T3 und T4.
→ 4) Kortikotrope Funktion: ACTH und Cortisol basal sind erniedrigt und nach Gabe von CRH bzw. im Insulin-Hypoglykämie-Test zeigt sich ein zu geringer Anstieg der Hormone.
→ 5) Laktotrope Funktionsüberprüfung erfolgt durch Gabe von TRH, die beim Panhypopituitarismus durch einen zu niedrigen Prolaktinspiegel auffällt.
→ IV: Bildgebung mit CT und MRT zur Tumorsuche bzw Ausschluss.
→ Klinisch-relevant:
→ A) Bei hypothalamischen Prozessen ist Prolaktin aufgrund der Beeinträchtigung des Prolaktin-inhibitin-factor (= Dopamin) eher erhöht.
→ B) Ein normaler TSH-Wert schließt eine sekundäre Hypothyreose bzw. Störung der thyreotropen Funktion nicht aus, sodass bei Verdacht auf HVL-Insuffizienz auch immer die fT4-Konzentration mitbestimmt werden sollte.
→ Differenzialdiagnose: Von der Hypophysenvorderlappen-Insuffizienz müssen insbesondere nachfolgende Erkrankungen abgegrenzt werden:
→ I: Polyglanduläres Autoimmunsyndrom: Auch hier zeigen sich endokrine Funktionsverluste, jedoch sind zumeist die NNR (im Sinne einer Nebennierenrinden-Insuffizienz) oder die Schilddrüse (Hashimoto-Thyreoiditis) betroffen.
→ II: Essstörungen: Bei der Essstörung, Anorexia nervosa, bildet sich überwiegend auch ein Hypogonadismus aus, jedoch ohne Verlust der sekundären Behaarung.
→ Therapie:
→ I: Medikamentöse Therapie: Hier steht die Gabe der fehlenden adenotropen Hormone im Vordergrund:
→ 1) ACTH-Mangel: Applikation von Hydrocortisol 20-30mg/d, wobei 2/3 der Dosis morgens, der Rest über den Tag verteilt verabreicht wird (20-5-5mg). Insbesondere bei Infektionen und Operationen kann eine Erhöhung der Tagesdosis auf das 2-3-fache nötig sein. Müdigkeit, Appetitlosigkeit oder Übelkeit weisen auf eine Unterdosierung hin. Kontrollparameter sind u.a. das Befinden des Patienten, der Blutdruck und die Serum-Natriumkonzentration.
→ 2) GH-Mangel: Bei Auftreten eines Wachstumshormonmangels im Erwachsenenalter ist eine Hormonsubstitution nicht nötig. Bei Kindern sollte vor Epiphysenfugenschluss die Therapie beginnen. Es erfolgt die Applikation eines synthetisch-hergestellten humanen GH mit einer Initialdosis von 0,15-0,3mg/d bis zu einer Maximaldosis von 1mg/d. Kontrollparameter sind der IGF-1 Wert sowie das Befinden des Patienten. Zudem hat die Wachstumshormonsubstitution positive Auswirkungen auf Knochendichte, Muskelmasse und Muskelkraft.
→ 3) Gonadotroper-Mangel: Männern wird 250mg Testosteron intramuskulär alle 3-4 Wochen bzw. 1000mg alle 3 Monate verabreicht. Frauen wiederum wird ein Östrogen-Gestagen-Kombinationspräparat unter gynäkologischer Kontrolle verabreicht. Kontrollparameter sind für beide Geschlechter u.a. Serumtestosteron, sekundäre Geschlechtsmerkmale, Libido, Potenz, etc.
→ 4) TSH-Mangel: Applikationstherapie mit L-Thyroxin einschleichend mit einer Anfangsdosis von 50µg/d und schrittweisen Aufdosierung um 25-50µg/d (30min vor dem Frühstück). Die Zieldosis liegt zwischen 100-150µg/d. Kontrollparameter sind nicht das TSH, sondern vielmehr das fT4 sowie nach dem Befinden des Patienten.
→ Klinisch-relevant: Die mittlere Tagesdosis von Hydrocortison muss in Belastungssituationen deutlich erhöht (5-10-fache) werden, um die Entwicklung einer Addison-Krise zu vermeiden.
→ II: Operative Therapie: Bei Tumoren erfolgt eine operative Therapie oder evtl. Bestrahlung mit anschließender Hormonsubstitution.