- Details
- Kategorie: Intensivmedizinische Krankheitsbilder des Herz-Kreislauf-Systems
- Zugriffe: 16867
→ Definition:
→ I: Bei der ventrikulären Tachykardie handelt es sich um 3 oder mehr unterhalb des His-Bündels entstehende Extrasystolen mit einer Frequenz zwischen 100-240 Schlägen/min. Charakteristikum hierbei ist ein verbreiterter QRS-Komplex (> 120ms), dessen Ursprung im Ventrikelmyokard liegt und über einen aberrierende Erregungsleitung verläuft.
→ II: Von einer anhaltenden ventrikulären Tachykardie spricht man, wenn sie länger als 30sec. anhält (bei weniger als 30sec. spricht man von einer nicht-anhaltenden Kammertachykardie).
→ Ätiologie: Ventrikuläre Tachykardien manifestieren sich v.a. bei ausgeprägten strukturellen Herzerkrankungen (z.B. Myokardinfarkt).
→ I: Bei schwere Herzerkrankungen, insbesondere chronisch-ischämische Prozesse wie die KHK und der Myokardinfarkt.
→ II: Genetisch bedingte Ionenkanalerkranungen des Herzens wie das Short-QT-Syndrom, Long-QT-Syndrom und das Brugada-Syndrom können eine VT einleiten.
→ III: Des Weiteren erworben bei Kardiomyopathien (z.B. dilatative Kardiomyopathie, arrhythmogene-rechtsventrikuläre Kardiomypathie), Myokarditis, schwere Rechtsherzbelastungen (z.B. beim Cor pulmonale oder Lungenembolie etc.), sowie medikamenteninduziert durch Digitalis oder andere Antiarrhythmika.
→ IV: Sehr selten tritt eine ventrikuläre Tachykardie bei Herzgesunden auf; sie stellt eine idiopathische ventrikuläre Tachykardie dar und wird in folgende Subtypen unterteilt:
→ 1) Idiopathische linksventrikuläre Tachykardie (ILVT),
→ 2) Rechtsventrikuläre Ausflusstrakttachykardie (RVOT),
→ 3) Linksventrikuläre Ausflusstrakttachykardie (LVOT).
→ Pathogenese:
→ I: Meist handelt es sich um Reentry-Kreisläufe im myokardialen Randgebiet von Narbengeweben bei KHK oder Myokardinfarkt. Hierbei kreist die Erregung in der Übergangszone zwischen der Infarktnarbe (= Erregungsbarriere) und vitalem Myokardgewebe (mit elektrophysiologisch abnormen bzw. verlangsamten Leitungseigenschaften). Einen weiteren Pathomechanismus stellt eine gesteigerte Autonomie, die insbesondere im Bereich des rechts- und linksventrikulären Ausflusstracktes lokalisiert ist, dar.
→ II: Getriggerte Aktivität infolge einer Digitalisintoxikation,
→ III: Selten durch abnorme Autonomien bei Herzgesunden (infolge idiopathischer, ventrikulärer Tachykardien).
→ Klassifikation: Die ventrikuläre Tachykardie kann nach Dauer und Morphologie klassifiziert werden:
→ I: Nach Dauer: Weniger als 30sec. = nicht-anhaltende, länger als 30sec. = anhaltende VT.
→ II: Nach Morphologie: Monomorphe VT mit uniformen QRS-Komplexen (= konstante QRS-Morphologie), polymorphe VT mit morphologisch wechselnden QRS-Komplexen.
→ Klinik: Je nach Frequenz, Dauer und Zustand des Herzkreislaufsystems können die Symptome stark variieren:
→ I: Kurz anhaltende VT können asymptomatisch verlaufen.
→ II: Länger dauernde entwickeln klinische Zeichen aufgrund der Abnahme des Herzminutenvolumens mit Kaltschweißigkeit, Palpitation, Herzrasen, Dyspnoe, Hypotonie, Angina pectoris bis hin zum Lungenödem und kardiogenem Schock.
→ Komplikationen:
→ I: Übergang in ein Kammerflimmern mit Ausbildung eines kardiogenen Schocks.
→ II: Beim Long-QT-Syndrom besteht die Gefahr der Entwicklung einer Torsades-de-Pointes-Tachykardie (EKG-Befund: Torsades-de-Pointes-Tachykardie).
→ Diagnose:
→ I: Anamnese: Medikamentenanamnese (z.B. Antiarrhythmika, Herzglykoside), Bestimmung des Digoxin/Digitoxin-Serumspiegels.)
→ II: EKG/LZ-EKG/Event-Recorder: Mittel der Wahl zum Nachweis einer ventrikulären Tachykardie:
→ 1) Deformierte, schenkelblockartig-verbreiterte QRS-Komplexe mit einer Dauer von zumeist > 120msec., meist > 140msec. (EKG-Befund: Schenkelblock allgemein).
→ 2) AV-Dissoziation, die P-Welle besteht unabhängig vom QRS-Komplex.
→ 3) Wenn möglich mit älteren EKGs vergleichen. (Siehe auch EKG-Befund: Ventrikuläre Tachykardien).

→ Differenzialdiagnose: Von der ventrikulären Tachykardie müssen insbesondere nachfolgende Erkrankungen abgegrenzt werden; hierzu zählen u.a.:
→ I: Supraventrikuläre Tachykardien mit Aberration,
→ II: Supraventrikuläre Tachykardie mit vorbestehendem Schenkelblock.
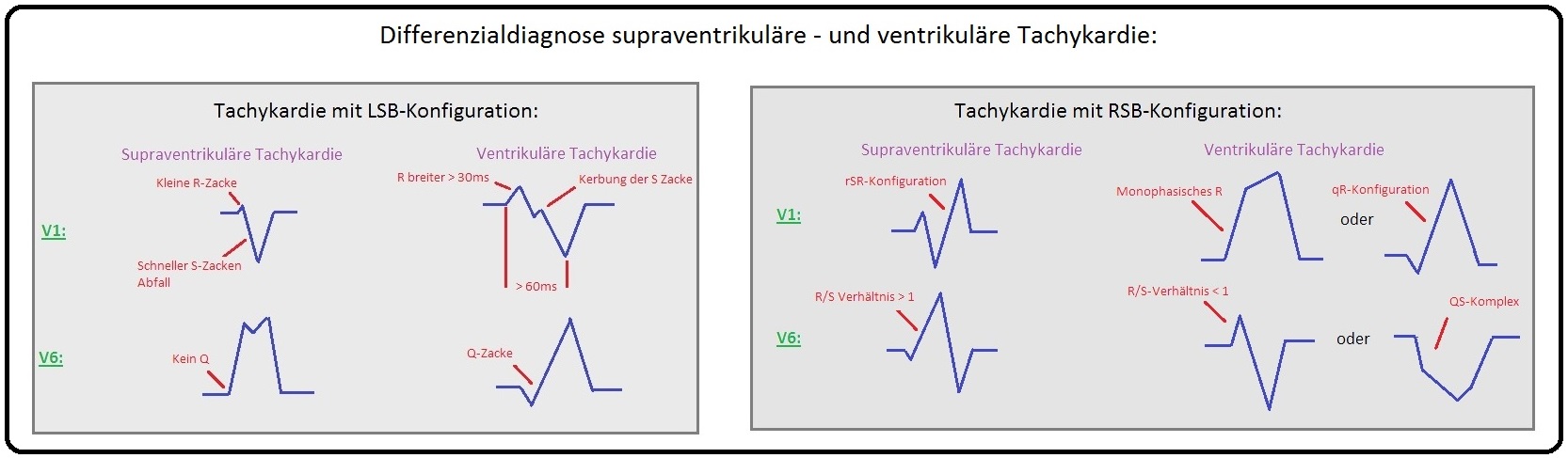
→ Klinisch-relevant: Alle Tachykardien mit breitem QRS-Komplex werden bis zum Beweis des Gegenteils als Kammertachykardie gewertet und stellen immer eine Notfallsituation dar.
→ Therapie:
→ I: Akuttherapie:
→ 1) Überprüfung einer Digitalisintoxikation und der Elektrolyte, insbesondere der Kalium-Serumkonzentration (Hypokaliämie/Hyperkaliämie) sowie die O2-Gabe.
→ 2) Medikamentöse Therapie:
→ A) Amiodaron: Stellt das Mittel der Wahl bei Patienten mit Herzinsuffizienz dar. Die Dosierung beträgt 300mg i.v. über 5min unter EKG-Kontrolle.
→ C) Ajmalin: Ist bei Patienten ohne Herzinsuffizienz das Mittel der Wahl mit einer Dosis von 50mg langsam i.v. unter EKG Kontrolle.
→ E) Zugleich sollte eine Korrektur der begünstigenden/auslösenden Faktoren wie Hypoxie, Elektrolytverschiebungen (z.B. Kalium-, Magnesium-Substitution), Azidose eingeleitet werden.
→ 3) Kardioversion: Bei hämodynamisch instabilen Patienten sollte eine sofortige elektrische Kardioversion unter Kurznarkose mit einer Initialdosis von 200J und anschießender Gabe von Amiodaron erfolgen.
→ 4) Bei bestehender Bewusstlosigkeit mit Kammerflattern/Kammerflimmern ist eine sofortige Defibrillation indiziert.
→ II: Rezidivprophylaxe:
→ 1) Behandlung der Grunderkrankung z.B. durch eine Revaskularisationstherapie bei bestehender Myokardischämie.
→ 2) Nach Myokardinfarkt ist eine Therapie mit ß-Blockern (ohne intrinsische Aktivität) indiziert, um das Risiko des plötzlichen Herztodes zu minimieren.
→ 3) Bei Patienten mit instabilen VT und/oder einer verminderten Ejektionsfraktion (< 35%) sollte ein ICD (= implantierbarer Kardioverter-Defibrillator) implantiert werden.
→ 4) Rezidivierende VT trotz ICD-Implantation bzw. antiarrhythmischer Therapie bedürfen einer Katheterablation.
→ Prognose: Die Prognose ist insbesondere von der zugrundeliegenden Erkrankung abhängig. Als prognostisch ungünstig erweist sich eine anhaltende ventrikuläre Tachykardie in den ersten 3 Monaten nach Myokardinfarkt; hierbei liegt die Mortalitätsrate bei bis zu 85% innerhalb des ersten Jahres. Patienten ohne organische Herzerkrankung haben kein relevant erhöhtes Mortalitätsrisiko.
- Details
- Kategorie: Intensivmedizinische Krankheitsbilder des Herz-Kreislauf-Systems
- Zugriffe: 28784
→ Definition: Beim Lungenödem handelt es sich um eine massive, extravaskuläre Flüssigkeitsansammlung (aus den Lungenkapillaren) initial im Lungenintersitium, später auch im Alveolarraum mit konsekutiver Behinderung des Gasaustausches. Hierbei unterscheidet man zwischen 2 Formen:
→ I: Dem interstitielle Lungenödem: Initial durch Flüssigkeitsansammlung im Bereich der Zellzwischenräume (bzw. des Lungengewebes) und
→ II: Dem alveolären Lungenödem: Im weiteren Krankheitsverlauf tritt die Flüssigkeit in den Alveolarraum (Alveolen, Bronchiolen) über.
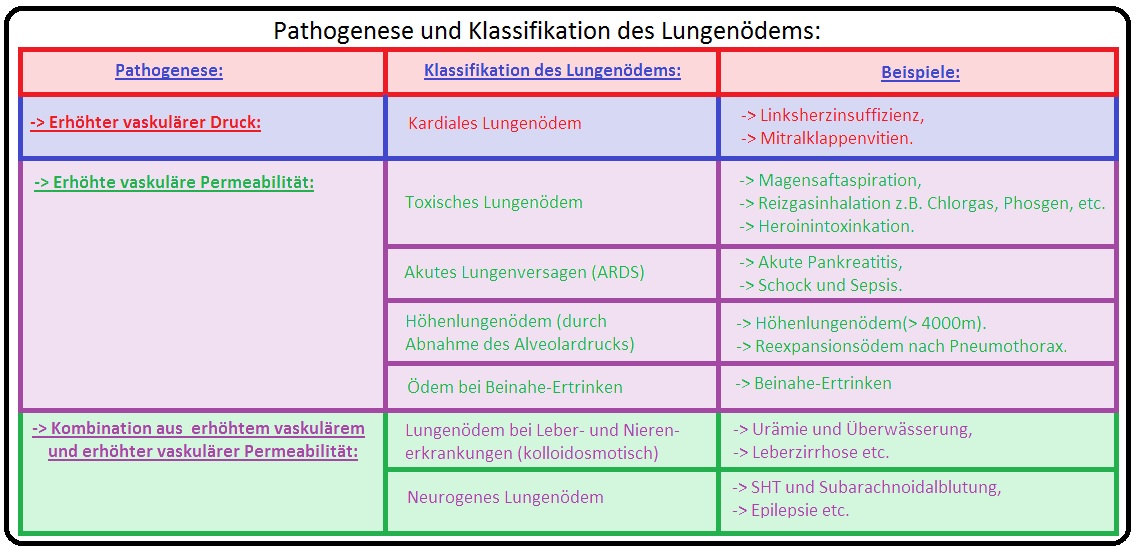
→ Ätiologie: Hinsichtlich der Genese wird es in einen kardialen- und einen nicht-kardialen Typ unterteilt:
→ I: Kardiales Lungenödem: Meist aufgrund einer akuten/chronischen Linksherzinsuffizienz mit immensem Druckanstieg im Lungenkreislauf, verursacht durch:
→ 1) Einen Myokardinfarkt,
→ 2) Hypertensive Krisen,
→ 3) Myokarditiden,
→ 4) Tachykarde und bradykarde Herzrhythmusstörungen oder
→ 5) Eine Dekompensation einer chronischen Herzinsuffizienz bzw. aufgrund von Herzklappenvitien, insbesondere die Mitralinsuffizienz und die Mitralstenose.
→ II: Nicht-kardiales Lungenödem:
→ 1) Durch Abfall des onkotischen Drucks bei akuter oder chronisch dekompensierter Niereninsuffizienz kommt es zum massiven Flüssigkeitsübertritt
→ 2) Erniedrigung des Alveolardrucks bei Punktion großer Mengen Flüssigkeit (Pleurapunktion von > 1,5l kann zu einem Postexpansions-Lungenödem führen).
→ 3) Permeabilitätssteigerung der Alveolargefäße aufgrund allergischer Reaktionen (anaphylaktischer Schock), Noxen wie Reizgase (z.B. Chlorgas), Heroinintoxikation, Magensaftaspiration (Mendelson-Syndrom) oder Aspiration von Süß- und Salzwasser.
→ 4) Weitere Ursachen: Sind Lungenembolie und Schädel-Hirn-Traumata, Komplikationen einer Rickettsiose, infolge einer Sympathikusaktivierung mit konsekutiver Vasokonstriktion.
→ Pathogenese: des kardialen Lungenödems:
→ I: Im Zuge der Linksherzinsuffizienz staut sich das Blut vor dem linken Ventrikel in den Lungenkreislauf zurück mit konsekutivem Druckanstieg. Übersteigt der hydrostatische Druck den entgegengesetzten Drücken, (nämlich Alveolardruck, Gewebedruck und onkotischer Druck) tritt Flüssigkeit aus den Gefäßen in das Lungengewebe (= Interstitium) und im weiteren Krankheitsverlauf in den Alveolarraum über.
→ II: Folgen: des Lungenödems sind u.a.:
→ 1) Abnahme der Lungencompliance und der Vitalkapazität,
→ 2) Erhöhung der Diffusionsstrecke für Sauerstoff,
→ 3) Anstieg des Atemwiderstandes und
→ 4) Entwicklung einer ausgeprägten respiratorischen Insuffizienz mit Hypoxämie (= Abfall der Sauerstoffsättigung im Blut).
→ Klassifikation: Das Lungenödem wird in 4 Stadien unterteilt:
→ I: Stadium I: Interstitielles Lungenödem mit Flüssigkeitsansammlung im Lungengewebe.
→ II: Stadium II: Alveoläre Lungenödem mit Exsudat bzw. Transsudat in den Alveolen und Bronchiolen.
→ III: Stadium III: Schaumbildung mit weiterer Flüssigkeitsausbreitung.
→ IV: Stadium IV: Asphyxie.
→ Klinik:
→ I: Interstitielle Lungenödem: Verschärftes Atemgeräusch mit Giemen, Dyspnoe, Tachypnoe, Orthopnoe evtl. Husten (Asthma cardiale).
→ II: Alveoläres Lungenödem: Körperliche Unruhe, Todesangst, schwerste Dyspnoe, Blässe, Zyanose, feuchte Rasselgeräusche, schaumiges Sputum, Schocksymptomatik. Insbesondere beim ausgeprägten alveolären Lungenödem kardialen Ursprungs kann aus der Distanz ein Trachealrasseln hörbar sein und nicht selten wird ein rötliches, seröses, z.T. schaumiges Sekret abgehustet.
→ Diagnose:
→ I: Anamnese: Kardiale Vorerkrankungen,
→ II: Klinische Untersuchung:
→ 1) Inspektion: kaltschweißige, blasse, evtl. zyanotische Haut, Halsvenenstauung
→ 2) Auskultation: Feuchte Rasselgeräusche initial feinblasig, später grobblasig, zumeist basal lokalisiert, rechts > als links.
→ III: EKG-Monitoring: Eventuell Herzrhythmusstörungen, Ischämiezeichen usw.
→ IV: Röntgen:
→ 1) Parahiläre, schmetterlingsförmige Verschattungen,
→ 2) Beim interstitiellen Lungenödem Nachweis von Karley-B-Linien = laterobasal lokalisierte, horizontal verlaufende Linien in beiden Lungenhälften.

→ 3) Beim alveolären Lungenödem diffuse Verschattungen = Milchglaszeichnung der Lunge.
→ 4) Evtl. radiologisch nachweisbare Zunahme der Herzgröße bei Linksherzinsuffizienz.
→ 5) Das interstitielle Lungenödem ist nur radiologisch (perihiläre schmetterlingsförmige Verschattungen) nachweisbar, das alveoläre Lungenödem imponiert auskultatorisch durch feuchte Rasselgeräusche.
→ V: Labor: Zu bestimmen sind BB, Elektrolytstatus, Retentionswerte (Harnstoff, Kreatinin), Marker der Myokardschädigung (CK, CK-MB,Troponin I/T, LDH etc.) und die BGA z.B. durch Punktion der A. radialis oder ulnaris (Abb.: Normwerte der BGA).
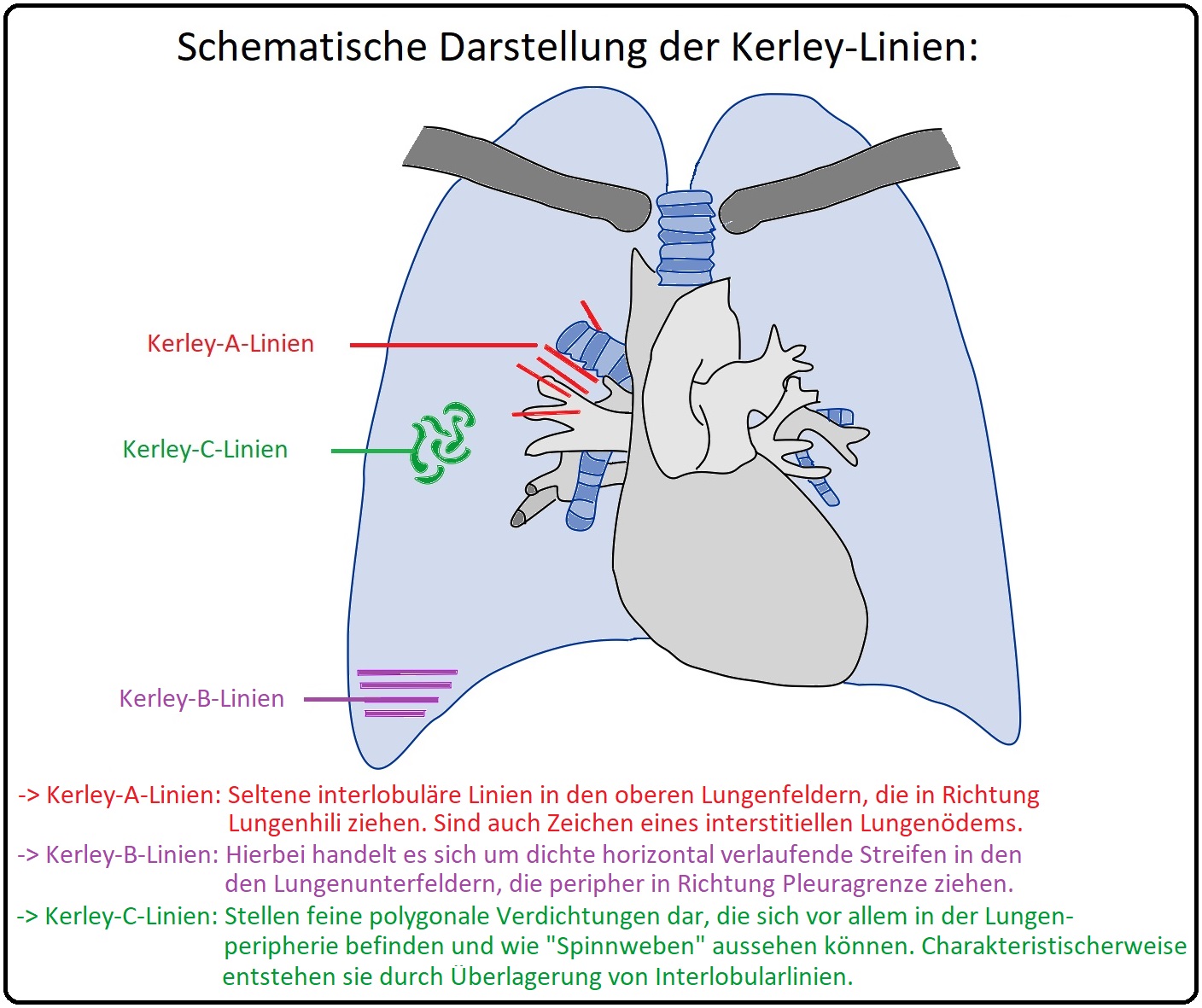
→ Klinisch-relevant: Die Kerley-Linien (= Kerley-A/-B/-C-Linien) sind im Röntgenbild transiente, maximal 1mm breite Linien, die durch eine ödematöse Erweiterung der Bindegewebssepten und der Lymphgefäße entstehen. Es werden hierbei 3 Typen von Kerley-Linien unterschieden:
→ A) Kerley-A-Linien: (= APIKAL) Radiologisch handelt es sich 4-6cm lange, irreguläre Linien, die in der Regel von den Oberlappen Richtung Lungenhili ziehen. Diese selten auftretenden Linien entstehen aufgrund einer Verdickung der Interlobulärsepten und treten u.a. beim interstitiellen Lungenödem auf.
→ B) Kerley-B-Linien: (= BASAL) Hierbei handelt es sich um 1-2cm lange, horizontal verlaufende, nicht-verzweigende Linien, die in den Lungenunterfeldern (= laterobasal) lokalisierte sind und in der Regel die pleurale Grenze erreichen. Sie können durch eine chronische pulmonalvenöse Drucksteigerung z.B. bei Mitralstenose oder Lymphangiosis carcinomatosa fibrosieren und folglich persistieren. Sie sind insbesondere im Bereich des Recessus phrenicocostalis gut erkennbar.
→ C) Kerley-C-Linien: (= CENTRAL) Sie stellen feinmaschige Netzzeichnungen dar, die diffus auf allen Lungenfeldern auftreten können, aber häufig zentral nachweisbar sind.
→ Differenzialdiagnose: Vom Lungenödem sind u.a. nachfolgende Erkrankungen abzugrenzen:
→ I: Die Pneumonie imponiert häufig durch Fieber und den einseitigen Befall der Lunge (Infiltration).
→ II: Das Asthma bronchiale ist insbesondere gekennzeichnet durch das Auftreten von:
→ 1) Trockenen Rasselgeräuschen,
→ 2) Trockener Haut und
→ 3) Durch den Nachweis einer pulmonalen Vorgeschichte.
→ III: Lungenembolie und
→ IV: Atemstörungen bei zerebralen Erkrankungen oder exogenen/endogenen Intoxikationen.
→ Therapie:
→ I: Allgemeinmaßnahmen: Sitzende Lagerung mit herabhängenden Beinen (herabhängende Beine vermindern den venösen Rückfluss und senken somit den hydrostatischen Druck in der Lunge). Die Oxygenierung erfolgt initial 4-8l über eine Maske (CAVE: Bei Patienten mit COPD), nachfolgend nach BGA; evtl. Intubation. Sedierende Maßnahmen (Todesängste) mit Morphin 3-5mg (1:10 verdünnt) s.c. und/oder i.v.
→ Klinisch-relevant: Morphin hat eine vasodilatatorische Wirkung und senkt somit den pulmonalen Gefäßwiderstand.
→ II: Kardiales Lungenödem: Hierbei steht die Vorlastsenkung im Vordergrund:
→ 1) Nitrate: Wie Nitroglycerin 2-3Hub (in höheren Dosen senkt es auch die Nachlast. Anschließend Nitroperfusor 1-6ml/h (1ml = 1mg) nach RR.
→ 2) Schleifendiuretikum: Furosemid 40-80mg i.v. evtl. Wiederholung nach 30min. Bei Patienten mit Niereninsuffizienz ist die Initialdosis deutlich erhöht (125-250mg).
→ 3) Nicht-medikamentöser unblutiger Aderlass durch Abbinden einzener Extremitäten mit einer Blutdruckmanschette.
→ III: Bei Hypotonie: Gabe von Dopamin oder Noradrenalin + Dobutatmin.
→ 1) Dopamin: 1 Amp/5ml entspricht 200mg. Die Dosierung ist 2-12ml/h.
→ 2) Noradrenalin: 1 Amp enthält 1mg. Perfusortherapie (5 Amp auf 45ml einer 0,9%iger NaCl-Lösung) mit einer Dosierung von 3-12 ml/h. Es verursacht eine Vasokonstriktion und eine Steigerung des HZV.
→ 3) Dobutamin: 1 Amp (250mg trocken) auf 50ml einer G-5%iger Lösung. Die Dosierung ist 2-12ml/h. Dobutamin steigert das HZV und hat fast keinen Einfluss auf den Gefäßtonus
→ IV: Bei Herzrhythmusstörungen: Wie den ventrikuläre Tachykardien mit Salvenbildung kann folgendes appliziert werden:
→ 1) Amiodaron: (Cordarex) in einer Dosierung von 150-300mg/i.v. als Bolus über 10 min (bzw. Perfusortherapie 2ml/h).
→ 2) Verapamil (Isoptin) intravenös mit einer Dosierung von maximal 100mg/d (2-10ml/h).
→ V: Allgergisch-toxisches Lungenödem:
→ 1) Bei diesen Formen des Lungenödems ist eine inhalative Glukokortikoid-Applikation mit 5Hub/10min bis zum Abklingen der Symptome indiziert.
→ 2) Bei schweren Formen ist ggf. eine intravenöse Glukokortikoid-Therapie nötig.
→ VI: Kausale Therapie der Grunderkrankung.
→ Klinisch-relevant: Gerade bei der Inhalation von Reizgasen kann das Lungenödem noch nach einer Latenz von 12 Stunden auftreten, sodass eine prophylaktische stationäre Überwachung immer indiziert ist.
- Details
- Geschrieben von: CF
- Kategorie: Intensivmedizinische Krankheitsbilder des Herz-Kreislauf-Systems
- Zugriffe: 9890
→ Definition: Bei der Torsades-de-pointes-Tachykardie handelt es sich um eine Sonderform der polymorphen ventrikulären Tachykardie. Sie ist durch das Auftreten von paroxymalen, polymorphen Kammerkomplexen charakterisiert, die mit wechselnder Amplitudenhöhe und -richtung um die isoelektrische Linie tänzeln.
→ Klinisch-relevant:
→ A) Charakteristischerweise ist bei dieser Herzrhythmusstörung die QT-Zeit verlängert.
→ B) Die Herzfrequenz liegt in der Regel zwischen 200-250 Schläge/min.
→ Ätiologie: Die Entwicklung des Torsades-de-pointes-Tachykardie steht im direkten Zusammenhang mit passageren oder persistierenden QT-Verlängerungen; sie können angeboren oder erworben sein:
→ I: Angeborene Form: Beim angeborenen Long-QT-Syndrom aufgrund von Ionenkanalerkrankungen sind bis heute 6 Gen-Defekte bekannt, die insbesondere den Kalium-Kanal, seltener den Natrium-Kanal (Long-QT3) betreffen. Es kann autosomal-dominant (z.B. Romano-Ward-Syndrom) oder autosomal-rezessiv (z.B. Jerwell-Lange-Nielson-Syndrom) vererbt werden.
→ II: Erworbene Form: Hier stehen vor allem Risikofaktoren, die mit einer Verlängerung der QT-Zeit assoziiert sind, im Vordergrund; hierzu zählen:
→ 1) Strukturelle Herzerkrankungen: Diese gehen mit einer Herzrhythmusstörung aufgrund z.B. einer Herzinsuffizienz, KHK, Kardiomyopathie, etc. einher.
→ 2) Elektrolytstörungen: Insbesondere die Hypokaliämie und Hypomagnesiämie, aber auch die Hypokalzämie.
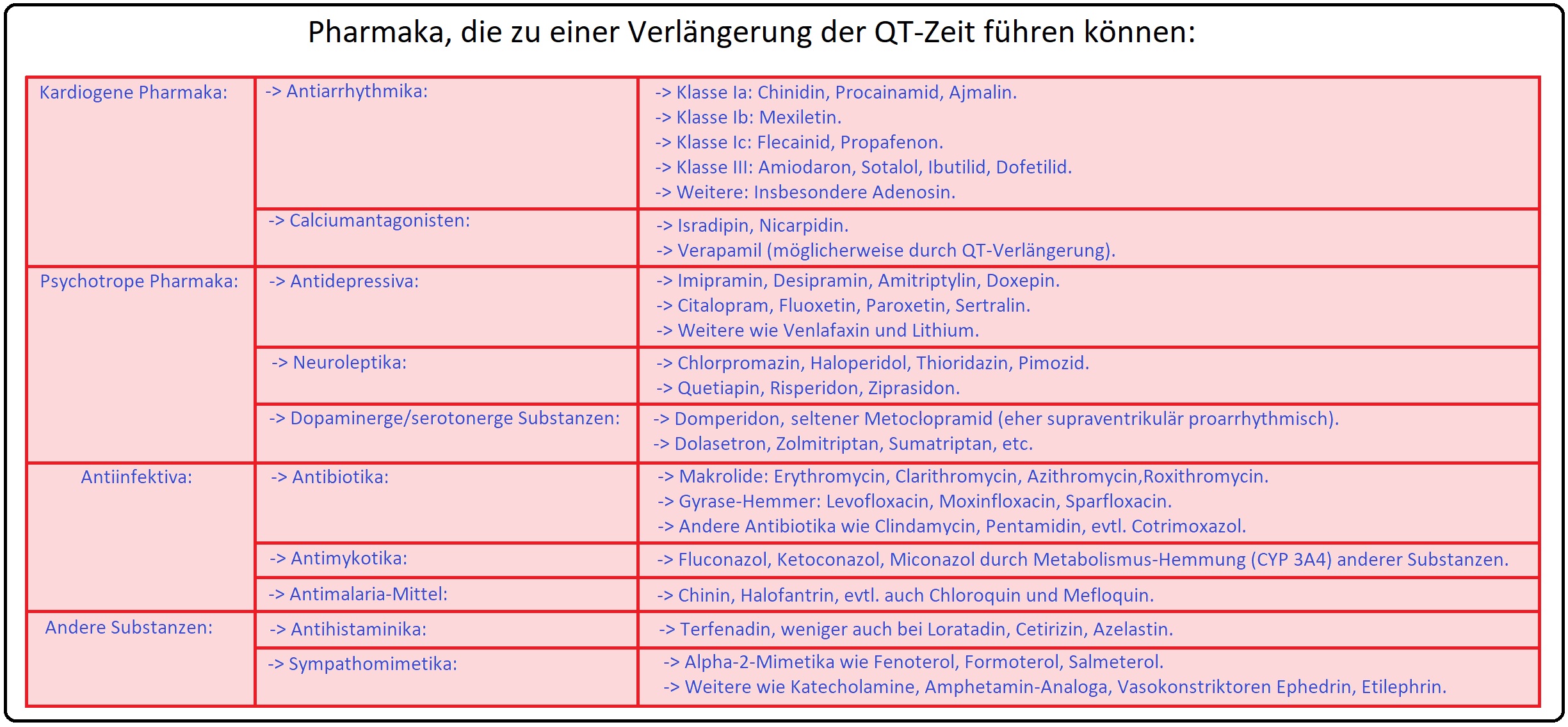
→ 3) Medikamenteninduziert: Bestimmte Medikamente induzieren einer Verlängerung der QT-Zeit und stellen somit einen wichtigen Risikofaktor für die Entwicklung einer Torsades-de-Pointes-Tachykardie dar:
→ A) Antiarrhythmika insbesondere die Klasse I (z.B. Chinidin) oder Klasse III (z.B. Sotalol, Amiodaron).
→ B) Antibiotika wie Fluorchinolone und Makrolide.
→ C) Antidepressiva, insbesondere die trizyklischen AD wie Doxepin und Amitriptylin und nicht zuletzt
→ D) Antipsychotika vor allem Pimozid, Ziprasidon, Haloperidol bei intravenöser Applikation, etc.
→ Pathogenese:
→ I: Die Repolarisationsphase des kardialen Aktionspotenzials wird durch den Kaliumausstrom und den Kalziumeinstrom in die Zelle bestimmt. Durch eine kongenitale oder erworbene Beeinträchtigung des Ionenausstausches, insbesondere des K+-Kanals, kann es zur Verlängerung der QT-Zeit mit konsekutiver Veränderung der Repolarisationsphase kommen.
→ II: Der genaue Pathomechanismus ist noch nicht genau geklärt, angenommen werden jedoch frühe Nachpotenziale, die in die vulnerable Phase der Repolarisation fallen.
→ III: Begünstigend für die Auslösung einer Torsades-de-Pointes Tachykardie sind u.a. die:
→ 1) Bradyarrhythmien insbesondere der AV-Block III Grades (EKG-Befund: AV-Block) sowie
→ 2) Spät einfallende Extrasystolen.
→ Klinik:
→ I: Die Torsades-de-pointes-Tachykardie tritt charakteristischerweise anfallsartig auf, insbesondere durch Stress getriggert und ist zumeist nur von kurzer Dauer.
→ II: Leitsymptome sind Palpitationen, Schwindel, Präsynkope und Synkope mit plötzlichem Bewusstseinsverlust.
→ III: Häufig ist diese Form der Rhythmusstörung selbstlimitierend und geht in ein Sinusrhythmus über; kann aber auch in ein Kammerflimmern mit plötzlichem Herztod degenerieren.
→ Diagnose:
→ I: Anamnese: Hierbei steht die Familienanamnese insbesondere die Eruierung eines plötzlichen Herztodes bei jüngeren Familienangehörigen sowie die Medikamentenanamnese im Vordergrund.
→ II: Labor: Elektrolytbestimmung mit besonderem Augenmerk auf die K+-, Na+-, Kalzium- und Magnesium-Plasmakonzentrationen.
→ III: EKG: Charakteristische EKG-Veränderungen sind u.a.:
→ 1) Verlängerung der QT-Zeit, insbesondere der frequenzkorrigierten QT-Zeit.

→ 2) Polymorphe, verbreiterte, den Vektor-wechselnde QRS-Komplexe.
→ 3) Unmittelbar vor Entstehung der Torsades-de-pointes Tachykardie nehmen die Repolarisationsstörungen zu und es zeigt sich im EKG eine charakteristische TU-Verschmelzung (EKG-Befund: Torsades-de-Pointes-Tachykardie).
→ IV: Echokardiographie: Zur Klärung von strukturellen Herzerkrankungen durch z.B. den Nachweis von Herzklappenveränderungen (Herzklappenvitien), Pumpfunktionsstörungen oder Wandbewegungsstörungen.
→ Differenzialdiagnose: Von der Torsades-de-pointes-Tachykardie müssen u.a. nachfolgende kardiale Erkrankungen abgegrenzt werden:
→ I: Synkopen anderer Genese wie z.B. die orthostatische Hypotonie,
→ II: Herzrhythmusstörungen: Wie
→ 1) Vorhofflattern und Vorhofflimmern
→ 2) Polymorphe bzw. multifokale ventrikuläre Tachykardien und das
→ 3) Kammerflimmern.
→ Therapie: Die zusätzliche Implantation eines ICD sollte bei der Torsades-de-pointes-Tachykardie immer erwogen werden.
→ I: Im Vordergrund der Behandlung der Torsades-de-pointes-Tachykardie steht die Therapie der Grunderkrankung, die Korrektur der Elektrolytentgleisung bzw. das sofortige Absetzten der auslösenden Medikamente.
→ II: Die Elektrolyt-Plasmaspiegel werden auf hochnormale Werte angehoben z.B, Kalium auf einen Zielwert von 4,8-5,2mmol/l durch Substitution von Kalium in einer Dosis von 5-15mmol/h i.v. oder langsame Gabe von Magnesiumsulfat 2g i.v. über 1-5min, anschließend 500mg/h i.v. bis auf einen Magnesium-Plasma-Zielwert von 0,9- 1,1mmol/l.
→ III: Bei Persistenz der Tachykardie bzw. Manifestation eines Kammerflimmerns ist eine elektrische Defibrillation mit 200-360J. indiziert. Evtl. muss die Implantation eines Herzschrittmachers (z.B. AAI) insbesondere bei bestehender Bradykardie erfolgen.
→ IV: Bei manifester kongenitaler Form ist bei Defekten des Kaliumkanals die Gabe eines ß-Blockers (z.B. Propranolol in hoher Dosis > 240mg/d), beim Natriumkanaldefekt ein Natriumkanalantagonist wie Mexiletin (600-720mg/d) oder Flecainid (100-200mg/d) indiziert.

→ Prognose:
→ I: Gerade bei Patienten mit bekannter struktureller Herzerkrankung muss auf Medikamente, die zu einer QT-Verlängerung führen, verzichtet werden.
→ II: Bei nicht bekannter Torsades-de-pointes-Tachykardie können sich rezidivierenden Synkopen und Kammerflimmern ggf. mit plötzlichem Herztod entwickeln.
- Details
- Kategorie: Intensivmedizinische Krankheitsbilder des Herz-Kreislauf-Systems
- Zugriffe: 24703
→ Definition: Bei der Lungenembolie handelt es sich um einen akuten Verschluss einer Pulmonalarterie, verursacht durch embolisch verschlepptes Material. In > 90% der Fälle wird sie durch einen Thrombus aus dem Versorgungsgebiet der Vena cava inferior (tiefe Beinvenenthrombose) hervorgerufen. Charakteristische Leitsymptome sind insbesondere u.a. akut auftretende Luftnot mit Tachypnoe, Thoraxschmerzen und evtl. Husten sowie Beklemmungsgefühl und Angst.
→ Epidemiologie:
→ I: Die Inzidenz liegt bei 60/100000/Jahr.
→ II: Ca. 10% der Patienten mit tiefer Beinvenenthrombose erleiden eine symptomatische Lungenembolie.
→ III: Eine bevorzugte Lokalisation ist der rechte Unterlappen (rechte Pulmonalarterie).
→ Ätiologie:
→ I: Die venösen Embolien stammen zum Großteil aus den Bein- und Beckenvenen bei evtl. bestehender Thrombophlebitis und Phlebothrombose, weniger aus den Armvenen und Uterusvenen.
→ II: Selten findet man Fettembolien aus den langen Röhrenknochen bzw. Luft- oder Fruchtwasserembolien.
→ III: Risikofaktoren, die eine Embolie begünstigen, sind:
→ 1) Immobilisation (Bettruhe, Flugreisen)
→ 2) Frühere TVT und Lungenembolien;
→ 3) Gerinnungsstörungen (Thrombophilie) wie AT-III-Mangel, Protein C und S-Mangel, APC-Resistenz insbesondere die Faktor-V-Leiden-Mutation, aber auch das Antiphopholipid-Syndrom, die paroxysmale nächtliche Hämoglobinurie, etc.
→ 4) Traumata und Operationen an der unteren Extremität und am Becken.
→ 5) Herzinsuffizienz;
→ 6) Schlaganfall sowie
→ 7) Maligne Erkrankungen und Polyzythämia vera.
→ 8) Weitere Faktoren: Sind Adipositas, Östrogen-Einnahme, Schwangerschaft, Zigarettenrauchen, etc.
→ Pathogenese: Der Embolus strömt mit dem venösen Blut in eine Pulmonalarterie. Sehr kleine Embolien werden durch das Fibrinolysesystem der Lunge eliminiert. Größere führen zu einer Verlegung des Pulmonalgefäßes mit Verminderung des Gesamtgefäßquerschnittes und konsekutiver Widerstandserhöhung im rechten Ventrikel (= Nachlasterhöhung). Folgen sind:
→ I: Akutes Cor pulmonale: Akute Rechtsherzbelastung mit Erhöhung des pulmonalarteriellen Mitteldrucks von 10 auf 30-40mmHg. Durch Freisetzung von vasoaktiven Mediatoren aus dem Embolus und Gefäßendothel entwickelt sich ein zunehmender Pulmonalarterienspasmus mit weiterer Widerstandserhöhung.
→ II: Infolge der begrenzten Kontraktilitätssteigerung des rechten Ventrikels entsteht eine Dilatation und Insuffizienz mit nachfolgender Abnahme des Herzminutenvolumens, Hypotonie bis hin zum kardiogenen Schock.
→ III: Ab dem Stadium III der Lungenembolie tritt eine Hypoxämie auf, die durch eine Zunahme des funktionellen Totraumes (hier besteht eine Ventilation ohne Perfusion) verursacht wird.
→ Pathophysiologische Komplikationen:
→ I: Lungenatelektasen: Sie können sich innerhalb von 24 Stunden durch Abnahme des Surfactant-Fakors ausbilden.
→ II: Hämorrhagische Lungeninfarkte: Stehen die Kollateralen (= Bronchialarterien) der Pulmonalarterien unter erhöhtem Druck bzw. Fehlen im Bereich der Segmentalarterien die Kollateralkreisläufe blutet es im Embolisationsbereich ins Gewebe ein. Folge ist ein hämorrhagischer Lungeninfarkt.
→ III: Gekreuzte Embolie: Durch die bestehende Obstruktion entwickelt sich eine pulmonal-arterielle Druckerhöhung mit Dilatation des rechten Ventrikels und Druckanstieg im rechten Vorhof. Hierdurch kann ein geschlossenes Foramen ovale wieder eröffnet werden, was über die Druckdifferenz zur Ausbildung eines Rechts-Links-Shunt führt. Der diesbezügliche Effekt ist, dass das embolische Material in den linken Vorhof und somit in den großen Kreislauf gelangt.
→ Phasen der Lungenembolie:
→ I: Phase 1: Obstruktion/Verschluss einer Pulmonalarterie mit rechtsventrikulärer Druckbelastung (Aferload erhöht)
→ II: Phase 2: Steigerung des funktionellen Totraumes mit arterieller Hypoxie und Myokardischämie und
→ III: Phase 3: Vorwärtsversagen mit Abnahme des HZV und kardiogenem Schock.
→ Klinik: Meist morgens nach dem Aufstehen, nach Defäkation sowie körperlicher Anstrengung mit:
→ I: Vegetativen Symptomen wie Angst, Beklemmung und Schweißausbruch.
→ II: Atemnot mit Dyspnoe und Tachypnoe.
→ III: Atemabhängigen Thoraxschmerzen, evtl. subphrenischem Projektionsschmerz
→ IV: Husten, Hämoptysen,
→ V: Synkopen und Schocksymptomatik.
→ Komplikationen: Sind insbesondere:
→ I: Pleuritis,
→ II: Hämorrhagischer Lungeninfarkt mit Hämoptysen,
→ 3) Infarkt-Pneumonie bzw. Abszessbildung,
→ 4) Rechtsherzversagen, pulmonale Hypertonie und Cor pulmonale.
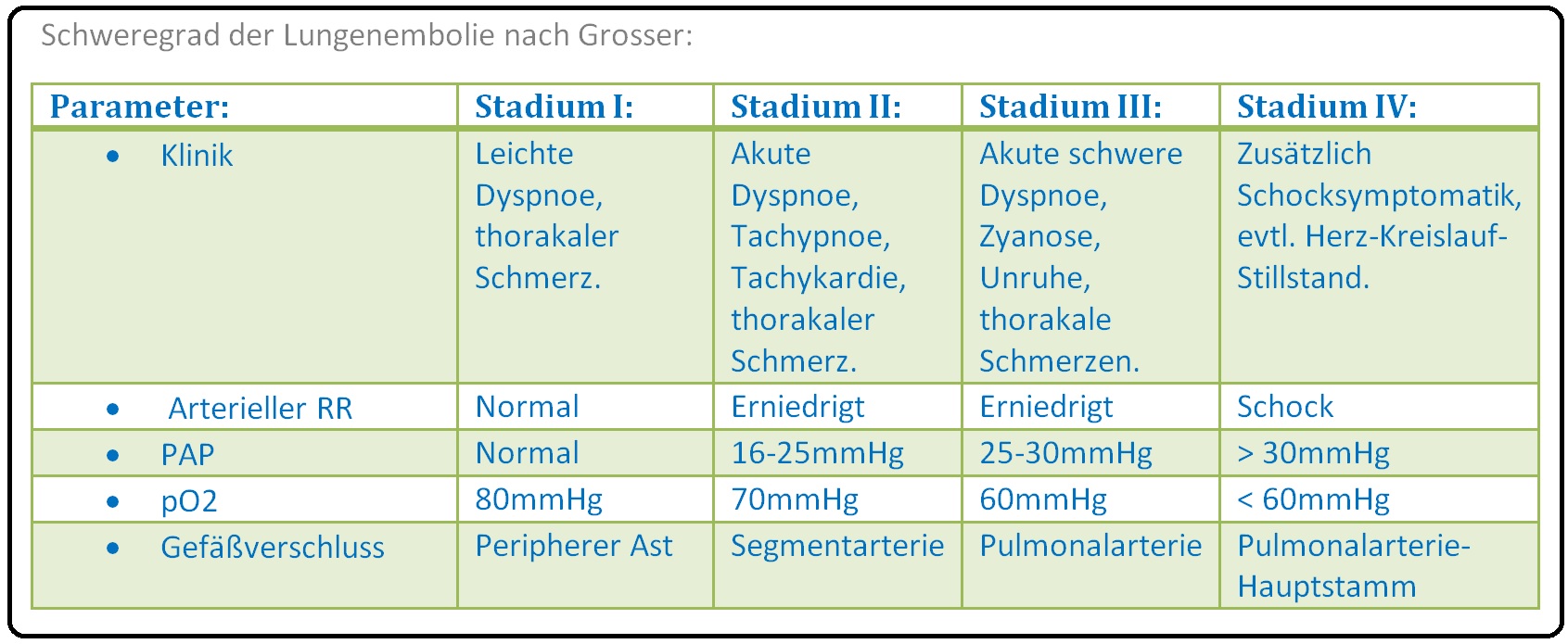
→ Stadieneinteilung: Nach Grosser:
→ I: Stadium 1: Das Stadium I ist klinisch evtl. durch leichte Dyspnoe und thorakale Schmerzen gekennzeichnet. Diagnostische Merkmale sind v.a. RR normal; pO2 > 75 mmHg, PAP (= pulmonalarterieller Mitteldruck) und normal < 20mmHg. Zumeist ist ein peripherer Ast obliteriert; die Letalität ist fast nicht vorhanden.
→ II: Stadium 2: Typische klinische Symptome sind Angst, akute Dyspnoe, Tachypnoe, atemabhängige Thoraxschmerzen, Tachykardie, evtl. Zyanose und Synkopen. Bezüglich der Diagnose sind normaler bis erniedrigt RR, normaler bis erniedrigter PO2 sowie ein PAP (pulmonalarterieller Mitteldruck) < 20mmHg (meist noch normal) nachweibar. Es handelt sich um einen Verschluss einer Segmentarterie, die Letalität liegt bei < 25% der Fälle.
→ III: Stadium 3:
Die Symptomatik gleicht der des Stadiums 2, z.T. treten Schockzeichen wie systolischer RR-Abfall < 100mmHg und Puls > 100/min auf. Zur Diagnosestellung müssen Kriterien wie deutlich erniedrigter RR, pO2 < 70mmHg und PAP (= pulmonalarterielle Mitteldruck) > 25-30mmHg eruierbar sein. Hierbei besteht ein Verschluss eines Pulmonalarterien-Astes die Letalität liegt > 25%.
→ IV: Stadium 4: Im Vordergrund der Klinik steht die ausgeprägte Schocksymptomatik z.T. reanimationspflichtig. Diagnostische Kriterien sind ein stark erniedrigt RR, pO2 < 60mmHg und PAP (pulmonalarterieller Mitteldruck) > 30mmHg. Es handelt sich um einen Verschluss des Pulmonalarterienstammes. Die Moralitätsrat ist mit > 50% der Fälle sehr hoch.
→ Diagnose:
→ I: Anamnese (insbesondere Eigenanamese, Vorerkrankungen, Familienanamese, etc.) sowie klinische Untersuchung (bei hämodynamisch instabilen Patienten sollte sofort ein ECHO bzw. Angio-CT erfolgen).
→ II: Labor:
→ 1) D-Dimere: (= D-Dimer-Antigen) Positiv bei akuter Embolie aber auch bei tiefe Venenthrombose und:
→ A) Z.n. Operationen (< 4 Wochen) und Traumata,
→ B) Gerinnungshemmender und fibrinolytischer Therapie sowie bei der disseminierten intravasalen Koagulopathie.
→ C) Schweren Entzündungen wie Erysipel, Pneumonie und Sepsis.
→ 2) Troponin I/T und BNP: Normale BNP-Wert und negatives Troponin I/T ist prognostisch günstig und schließt eine schwere Verlaufsform der LE aus.
→ 3) Blutgasanalyse: (Abb.: Normwerte der BGA) Die Diagnose der Lungenembolie wird erschwert bei kardiopulmonalen Vorerkrankungen. Man findet eine verminderte O2- und CO2-Konzentration.
→ 4) Die Gerinnungsaktivität (pTT und TZ) ist vermindert.
→ Klinisch-relevant:
→ A) Ein pO2-Wert > 80mmHg (schließt eine schwere Verlaufsform der LE aus.
→ B) Bei schwere Lungenembolie (Stadium III) kann eine arterielle Hypoxämie durch Oxygenierung nur geringfügig ausgeglichen werden.
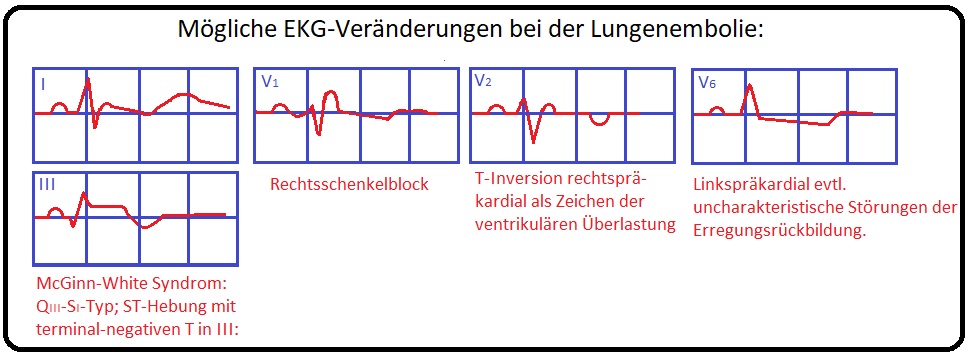
→ III: EKG: Siehe EKG-Befund Lungenembolie.
→ IV: Röntgen: Oftmals finden sich keine radiologischen Veränderungen. Mögliche radiologische Zeichen sind:
→ 1) Prominente zentrale Pulmonalarterie, evtl. Kalibersprung und Arterienlücke.
→ 2) Evtl. akut Herzvergrößerung
→ 3) Einseitiger Zwerchfellhochstand.
→ 4) Atelektasen als periphere keilförmige Infiltrationen
→ 5) Evtl. lokaler einseitiger Pleuraerguss.
→ 6) Charakteristische Hampton´s hump. Hierbei handelt es sich um alveoläre Hämorrhagien, die sich typischerweise bei der Lungenembolie entwickeln. Es handelt sich um halbkugelige periphere Verschattungen, die der Pleura aufsitzen und mit einer Latenz von bis zu 4 Tagen (frühstens nach 24 Stunden) nach dem Ereignis nachweisbar sind. Meist besteht begleitend noch ein einseitiger Pleuraerguss. Sie können sich innerhalb von einigen Tagen zurückbilden oder in einen Lungeninfarkt übergehen.
→ 7) Westermark-Zeichen: Zeichen lokaler Minderperfusion bei Hyperperfusion nicht betroffener Areale bei massiver LE.
→ V: Echo: Nicht sehr sensitiv. Nachweis von direkten und indirekten Zeichen einer LE mit:
→ 1) Bei Verschluss der Pulmonalarterie von > 30% = Nachweis von rechtsventrikulären Funktionsstörungen.
→ 2) Dilatation und Hypokinesien des rechten Ventrikels,
→ 3) Paradoxe diastolische Kammerseptumbewegung in den linken Ventrikel.
→ VI: CT-/MR-Angiographie: Mittel der 1. Wahl mit guter Darstellung der Arteria pulmonalis und deren Segmentarterien.
→ VII: Lungenperfusionsszintigraphie:
→ 1) Mit 99-Technetium markierten Albuminmakroaggregaten.
→ 2) Weist eine hohe Sensitivität auf; bei einem unauffälligen Befund kann eine Embolie fast ausgeschlossen werden.
→ 3) Die Embolie zeigt einen der Pulmonalarterie entsprechenden segment-spezifischen Ausfall der Lungenperfusion.
→ Differenzialdiagnose: Von der Lungenembolie müssen insbesondere nachfolgende Erkrankungen abgegrenzt werden:
→ I: Kardial: Myokardinfarkt, Angina pectoris, Herzrhythmusstörungen, Perikardtamponade,
→ II: Pulmonal/thorakal: Pneumothorax, Spannungspneumothorax, Asthma-bronchiale-Anfall, Lungenödem.
→ III: Nach Symptomen:
→ 1) Akute Luftnot: Pneumothorax, Lungenödem, Asthma bronchiale, Pneumonie.
→ 2) Akute Thoraxschmerzen: Angina pectoris, Myokardinfarkt, Aortendissektion, akutes Abdomen, Pankreatitis, Interkostalneuralgie, etc.
→ 3) Tachykardien: Supraventrikuläre Tachykardien, vasovagale Reaktion,
→ 4) Synkopen: Hypoglykämie, zerebrale Krampfanfälle, Intoxikation,
→ 5) Schock: Myokardinfarkt, rupturiertes Aortenaneurysma, bradykarde und tachykarde Rhythmusstörungen, Sepsis, Myokarditis.
→ Therapie:
→ I: Allgemein:
→ 1) Bei Verdacht auf eine Lungenembolie ist eine i.m. Injektion immer kontraindiziert.
→ 2) Oberkörperhochlagerung und Transport des Patienten wie ein rohes Ei, um weiteren Embolien zu vermeiden.
→ 3) Oxygenierung durch Gabe von 2-6 l/min O2 über eine Maske.
→ 4) Kontrolle der Vitalfunktionen mittels EKG, RR-Messung, Pulsoxymetrie.
→ 4) Evtl. Volumensubstitution mit einer kolloiden Lösung bei Hypotonie.
→ II: Medikamentös:
→ 1) Sedierung/Analgesie: Morphin (2,5-5mg i.v.), Diazepam 5-10mg/i.v.
→ 2) Katecholamine: Besteht eine Hypotonie bzw. Schocksymptomatik wird Dobutamin in einer Dosierung von 4-8µg/kgKG/min appliziert, bei massiver Embolie ist Noradrenalin (2-20µg/kgKG/min) indiziert.
→ 3) Heparinisierung: Heparin 5000-10000 IE i.v. als Bolus. Nachfolgend 1000-2000 IE/h (Zielbereich ist eine 1,5-2,5-fache Verlängerung des PTT Ausgangswertes), insbesondere bei GFR < 15-30 ml/min.
→ 4) Enoxaparin: (= Claxane) Niedermolekulares Heparin mit einer Dosierung von 2x 1mg/kgKG s.c. (bei GFR < 20-30 ml/min 1x täglich).
→ 4) Cumarine: Ab dem 1. oder 2. Tag erfolgt eine simultane Therapie mit Marcumar, deren Wirkung meist erst nach 3-5 Tagen einsetzt. Die Dauer der Marcumar-Therapie nach Lungenembolie ist wie folgt festgelegt:

→ 5) Das Heparin wird erst abgesetzt, wenn der INR-Wert an 2 aufeinanderfolgenden Tagen > 2-3 ist.
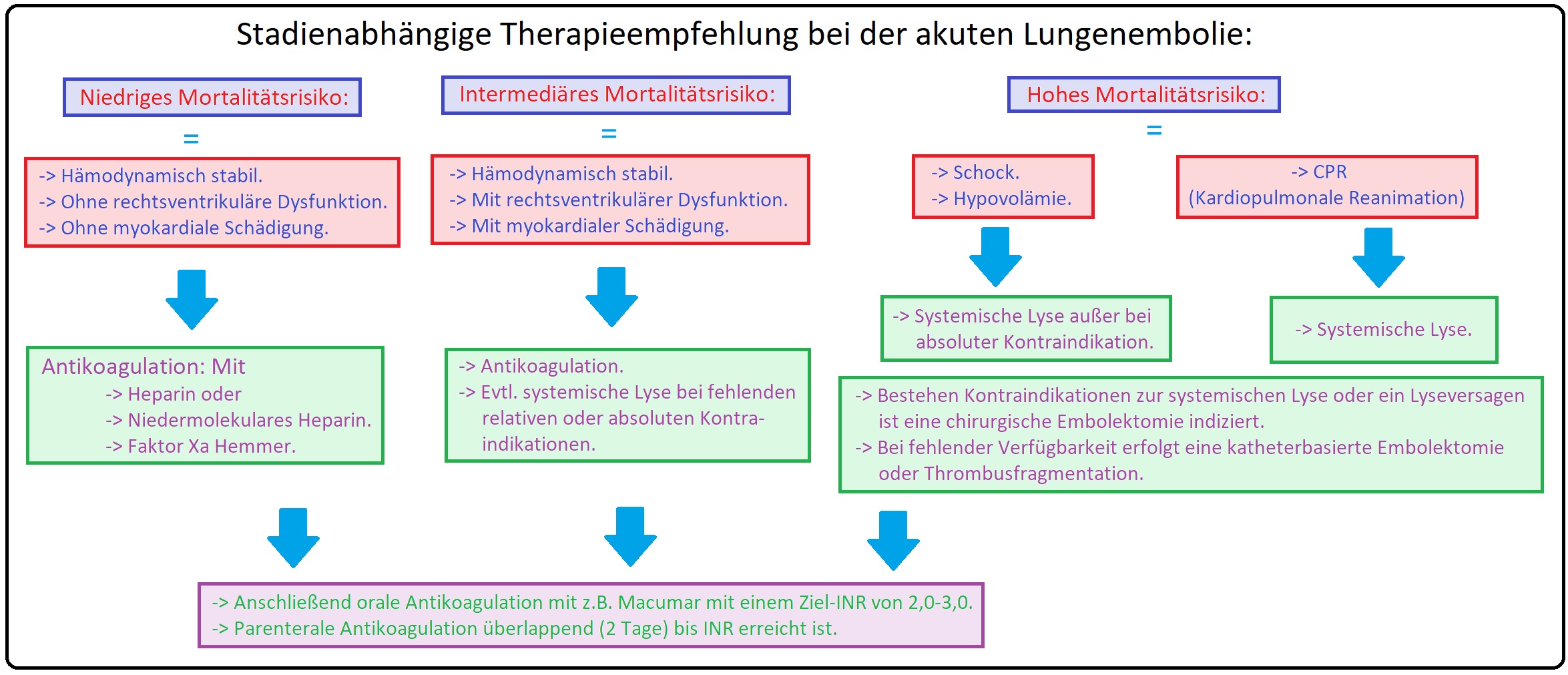
→ Klinisch-relevant:
→ A) Bei Fehlen von Kontraindikationen (wie Heparinallergie, heparininduzierte Thrombozytopenie Typ 2, bestehende hämorrhagische Diathesen, manifeste Blutungen, frische Hirninfarkte, Ösophagusvarizen etc.) ist Heparin das Mittel der Wahl zur Prophylaxe weiterer Embolie und senkt somit das Letalitätsrisiko.
→ B) Hierbei unterscheidet man:
→ 1) Unfraktioniertes Heparin: Indiziert bei hämodynamisch-instabilen, Patienten mit Niereninsuffizienz.
→ 2) Niedermolekulares Heparin: Clexane bei allen anderen Patienten.
→ III: Spezielle Therapie:
→ 1) Thrombolyse: Auflösung eines Embolus durch Fibrinolyse; es ist insbesondere indiziert bei hämodynamisch-instabilen Patienten und im Stadium III/IV nach Grosser. Die Durchführung beinhaltet:
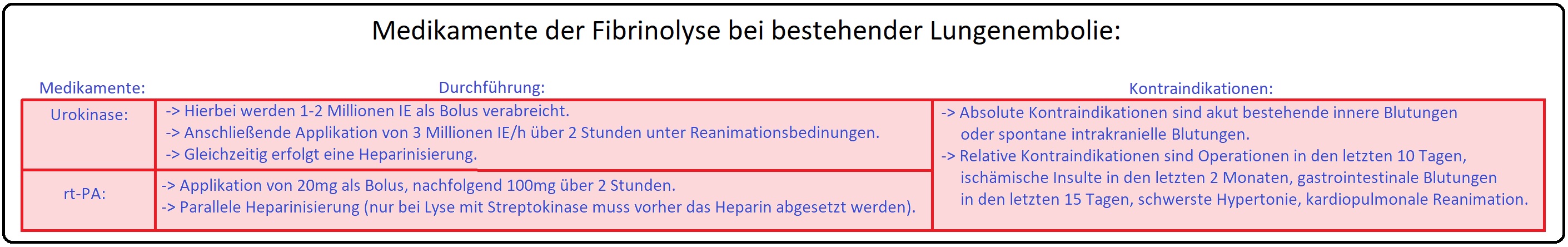
→ 3) Katheterfragmentation: Indiziert bei Kontraindikation gegen eine Fibrinolyse, bei Patienten im Stadium III/IV. Bei diesem Therapieverfahren wird das thrombotische Material mit Hilfe eines (Rechtsherz-) Katheters mechanisch zerkleinert.
→ 4) Operation: Die operative Embolektomie nach Trendelenburg ist bei massiver Lungenembolie mit hämodynamischer Instabilität und fehlendem Thrombolyseerfolg indiziert. Hierbei besteht ein hohes Letalitätsrisiko.
→ 5) Cava-Schirm: Einlage eines Vena-Cava Schirms bei rezidivierenden Lungenembolien und ineffektiver Antikoagulation. Die Schirmimplantation erfolgt unterhalb der Nierenarterien. Es besteht ein hohes Komplikationsrisiko durch Verschluss der Vena cava inferior.
→ Prognose:
→ I: Bei der Lungenembolie liegt die Rezidivrate bei 30%.
→ II: Die Prognose ist abhängig von:
→ 1) Dem Alter des Patienten,
→ 2) Bestehenden Vorerkrankungen,
→ 3) Schweregrad der Embolie und nicht zuletzt vom
→ 4) Zeitpunkt der Diagnosestellung bzw. adäquaten Therapiebeginn.